
Abstract
Williams syndrome is a neurodevelopmental disorder characterized by hypersociability and unique neurocognitive abnormalities. One of the characteristics of Williams syndrome is an inappropriate increase in social behavior. People with the syndrome may be overly friendly, even to strangers. We performed a novel study in our lab that demonstrated that the lack of a gene called Gtf2i may play a role in Williams syndrome. When this gene is absent, there are changes to one of the most important brain components for transferring signals in the brain and are likely to contribute to the over-friendliness observed in Williams syndrome. We expose surprising mechanisms involved in an increased social behavior, which is one of the characteristics of Williams Syndrome. In this article, we demonstrate how drugs can compensate for the damaged part of the brain and can bring the increased friendly behavior down to a normal level.
What is Williams Syndrome?
Imagine walking down the street and suddenly running into a dangerous person. You would most likely be startled, cross to the other side of the street, and possibly even call the police. Well, a person with Williams syndrome would probably be so excited to see a stranger’s face that he or she might not notice that the stranger is dangerous. People with Williams syndrome are described as having exceptionally friendly personalities, extremely sensitive and empathic, and therefore are also called “love children.”
Williams syndrome is a genetic disorder caused by the deletion of one of the two copies of about 26 genes found on chromosome 7 in humans [1]. Williams syndrome’s physical symptoms include problems with the heart and circulatory systems, hormonal issues, and unique facial features [1]. The behavioral symptoms of the syndrome include over-friendliness, increased empathy (the ability to understand the feelings of others), cognitive (mental) disorders, highly developed language skills, disrupted ability to recognize danger in social situations, and high levels of non-social stress (stress that is not related to social situations).
For many years, researchers have tried to decipher the mechanism behind Williams syndrome [1, 2]. Early studies found that one of the deleted genes in the syndrome, called Gtf2i, was related to the over-friendliness observed in Williams syndrome [3]. But the mechanism behind this gene influences excessive friendliness was not fully understood until more recently.
The Role of the Gtf2i Gene in Williams Syndrome
Different genes are expressed in different types of cells, and it is possible for scientists to silence a chosen gene by erasing it from certain cells or even from the whole body. In this study, we intentionally deleted the Gtf2i gene from nerve cells in the frontal brains of mice. We found that the model mice with deleted Gtf2i showed abnormal behaviors similar to those seen in Williams syndrome, including damage to the brain’s structure, gentle motor skills, and increased levels of friendliness and non-social stress (Figure 1).
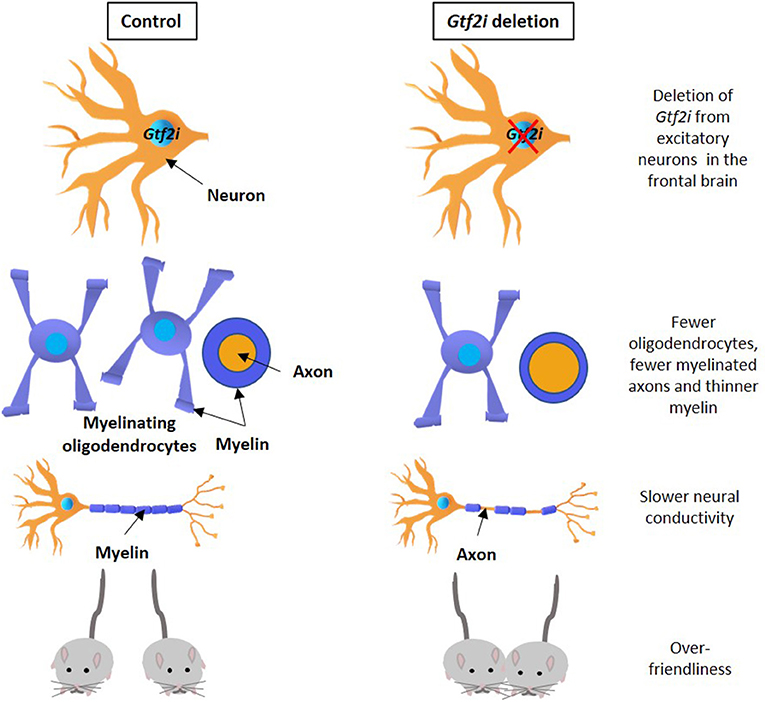
- Figure 1 - Gtf2i gene deletion and its consequences.
- Gtf2i gene deletion from nerve cells in the frontal brain of a mouse (right) causes damage similar to that seen in Williams syndrome, compared to control mice (left). The damage includes fewer oligodendrocytes, fewer axons covered in myelin and a thinner myelin layer (second row), lower neural conductivity (third row), and over-friendliness (bottom row). Adapted from Osso and Chan [4].
Through various experiments, we found a direct connection between the deletion of the Gtf2i gene in mice and issues with a substance called myelin. Myelin is a fatty layer covering and insulating the extensions of nerve cells, called axons. Without myelin, electrical signals pass through axons improperly and damage occurs in the central nervous system, leading to a functional problem, which could express for example in inadequate behaviors. This finding, which shows the influence of Gtf2i gene deletion on myelin, opens a new door for finding new treatments to Williams syndrome.
Results
Gtf2i Deletion in Nerve Cells in the Frontal Brain Results in Changes in Structural and Behavioral Changes
People with Williams syndrome show decreases in the volume of the brain and a thinner cortex (the outer layer of the brain). We wanted to check whether erasing the Gtf2i gene from nerve cells in the frontal brain is enough to cause those structural problems. The weight of the brain and the thickness of the brain’s cortex were significantly lower in 1 month-old mice without Gtf2i compared to control mice with the Gtf2i gene.
Additionally, our model mice showed significantly higher levels of social behavior, which we evaluated using three tests. Mice without Gtf2i had closer, more frequent, and longer interactions with other mice compared to control mice with the Gtf2i gene. We should also mention that our model mice preferred creating more interactions with mice that were “strangers” than with an inanimate object compared to control mice. We interpreted the behavior of model mice as a preference for performing increased social behavior.
Since one of the characteristics of Williams syndrome is a high level of non-social stress, we tested the mice for behaviors related to non-social stress and found significantly higher levels of non-social stress in our model mice compared to control mice.
Gtf2i Deletion Affects Myelin and Other Genes Related to Myelin
Although we deleted only the Gtf2i gene in the neurons of our model mice, we found that other genes were affected as well. We think that is because the protein made from the Gtf2i gene (Figure 2), called Tfii-I, is involved in regulating the expression of other genes. Surprisingly, 70% of the genes with lower expression in the cortex of 1-month-old Gtf2i-deleted mice were involved in the development and function of myelin or in the development and function of oligodendrocytes, which are the cells that produce myelin. We also found that the number of oligodendrocytes significantly decreased in the cortex of our model mice compared to control mice.
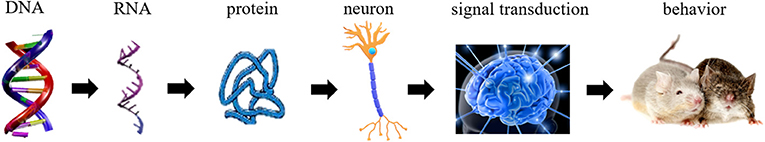
- Figure 2 - The chain of processes in the brain that eventually affects behavior.
- The genes in the DNA are transcribed to RNA, which is then translated into proteins. Proteins affect the activity of brain cells, including nerve cells, therefore affecting the way signals are sent in the brain. The way information is transferred in the brain affects behavior.
Gtf2i Deletion Causes Damage in the Myelin Structure, Neural Conductivity, and Motor Skills
In our model mice, we saw a significantly lower percentage of axons covered in myelin, and a significantly thinner layer of myelin on the axons, compared to control mice. This damage to the myelin layer affects neural conductivity, which is the way signals are sent along the axons of nerve cells.
Additionally, people with Williams syndrome show a decrease in fine motor skills. Our model mice also showed a lack of fine motor skills, along with weaker muscular tone in their front and rear limbs compared to control animals. This means that the damage to myelin influenced the motor skills of our model mice.
Fixing Damaged Myelin and Neural Conductivity Improves Neural and Behavioral Problems
We saw that, as a result of the myelin damage, our model mice had problems with neural conductivity. There is a drug called 4-AP that is known to improve neural conductivity. We gave this drug to our model mice and control mice to test whether it helped mice lacking Gtf2i improve their neural conductivity and, as a result, their motor skills and social behavior. We found that a single dose of 4-AP improved motor skills in our model mice. Additionally, model mice showed proper social preference in the social preference test. So, we found that treatment with 4-AP normalized the neural conductivity, therefore improving the social behaviors and motor skills in the model mice (Figure 3).
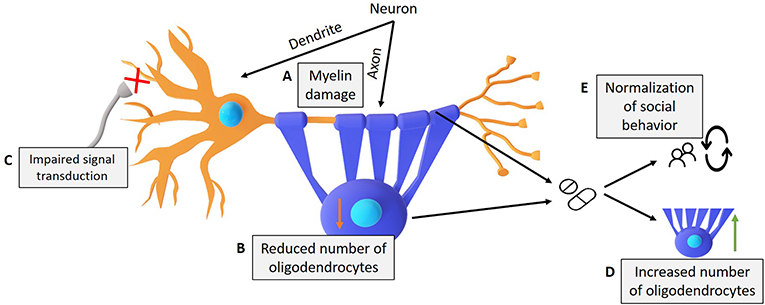
- Figure 3 - Damage that occurs in Williams syndrome, and drugs that might help.
- (A) Erasing the Gft2i gene from nerve cells results in damage to the myelin layer and (B) the number of oligodendrocytes (purple), which leads to (C) damage in the transfer of electrical signals. (D) We showed that the drug Clemastine increased the number of oligodendrocytes and fixed social behavior. (E) The drug 4-AP improved neural conductivity and fixed social behavior in model mice compared to control mice.
We also wanted to test whether fixing the damaged myelin could improve the social behaviors of our model mice. To test that, we used a drug called Clemastine to 1-month old model mice. The Clemastine treatment normalized the number of oligodendrocytes in the cortex and increased the thickness of the myelin layer in our model mice (Figure 3). We found that fixing the damage to the myelin was also sufficient to fix the increased social behaviors.
Since our studies were done in mice, we wanted to test whether our findings were also valid in people with Williams syndrome. We studied brain samples of frontal brain cortex from people with Williams syndrome who donated their brains to science and compared them to samples from a control group. We found a significantly lower number of oligodendrocytes and a sharp decrease in the thickness of the myelin layer in the frontal cortices of people with Williams syndrome compared to the control group, similar to our findings in mice. We also found that 19% of the genes that were expressed differently in people with Williams syndrome compared to the control group were related to myelin. Most of these genes are the same genes that were affected in our model mice.
Conclusions
By selectively erasing Gtf2i from nerve cells in mice, we found an unexpected decrease in the expression of genes related to myelin, in the number of oligodendrocytes, and in neural conductivity. We also saw similar results in tissues from humans with Williams syndrome. These data indicate that damage to myelin plays an important role in Williams syndrome, and that Gft2i deletion from neurons is probably the cause of this damage. This study identifies an important role of Gft2i expression in nerve cells, a crucial role for the development of oligodendrocytes and keeping proper myelin, and that the absence of these gene could contribute to some of the symptoms related to Williams syndrome.
This study sheds light on the role of myelin and neural conductivity in the inappropriate social behavior seen in Williams syndrome, and could therefore assist the development of new treatment strategies for behavioral disorders. Since the efficiency and speed of neural conductivity are dependent on myelin, dysfunctions in the features of myelin could affect the proper communication and synchronization between different brain areas crucial for performing adequate behaviors. Therefore, focusing on fixing myelin dysfunctions could be an efficient treatment strategy for Williams syndrome.
Glossary
Gtf2i: ↑ A gene that is missing in people with Williams syndrome. The absence of Gtf2i is correlated with the social disorders of the syndrome.
Model Mice: ↑ Mice that have been genetically modified serve as a model for a certain human disease. In this case, our model mice lacked the Gtf2i gene in neurons in the front of the brain.
Myelin: ↑ A fatty layer covering the axon to keep the signal transfer fast and efficient, much like an insulating material covering an electrical wire.
Axon: ↑ A long arm or extension of a nerve cell that carries the signal to the next cell.
Control Mice: ↑ Normal, healthy mice without genetic mutations, to which the model mice are compared.
Neuron: ↑ A nerve cell. The job of neurons is to receive electrical and chemical signals and transfer them to other nerve cells.
Oligodendrocytes: ↑ The cells that produce myelin.
Neural Conductivity: ↑ The way signals are sent along the axons of nerve cells.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Noa Schuchman and Roey Shemesh, exceptional young people who worked hard to process and edit the original article to suit everyone, took part in writing this article. Noa and Roi took an active part in various projects in Dr. Boaz Barak’s lab as part of the Alpha program of Tel Aviv University for Youth. Noa, 17, is studying chemistry and biotechnology at Ohel Shem High School in Ramat Gan. Roi, a 16-years-old from Petah Tikva, attends a gifted class at Ehad Ha’am High School, joined the Alpha program out of a great love for biology and the study of the human body.
Noa and Roi, we thank you from the bottom of our hearts and wish you success in the future.
Thank you!
Original Source Article
↑Boaz, B., Zhang, Z., Liu, Y., Nir, A., Trangle, S. S., Ennis, M., et al. 2019. Neuronal deletion of Gtf2i, associated withWilliams syndrome, causes behavioral and myelin alterations rescuable by a remyelinating drug. Nat. Neurosci. 22:700–8. doi: 10.1038/s41593-019-0380-9
References
[1] ↑ Pober, B. R. 2010. Williams-Beuren syndrome. N. Engl. J. Med. 362:239–52. doi: 10.1056/NEJMra0903074
[2] ↑ Morris, C. A., Kimberley, K. W., and Stock, A. D. 2003. GTF2I hemizygosity implicated in mental retardation in Williams syndrome: genotype-phenotype analysis of five families with deletions in the Williams syndrome region. Am. J. Med. Genet. A 123A:45–59. doi: 10.1002/ajmg.a.20496
[3] ↑ Barak, B., and Feng, G. 2016. Neurobiology of social behavior abnormalities in autism and Williams syndrome. Nat. Neurosci. 19:647–55. doi: 10.1038/nn.4276
[4] ↑ Osso, L. A., and Chan, J. R. 2019. A surprising role for myelin in Williams syndrome. Nat. Neurosci. 22:681–3. doi: 10.1038/s41593-019-0368-5