
摘要
年轻时,我痴迷于科幻小说,梦想着成为一名宇航员,我总是会被那些为世界带来技术革新的风云人物所吸引。 当我开始自己的科学生涯时,我希望在一些真正与众不同、有影响力的事情上施展拳脚,而不是被束缚在一些微不足道的小事上。这就是为什么我选择研究光学显微技术领域中最为棘手的问题之一——如何看到尺寸小于可见光波长的物体。这意味着我要挑战一个长期以来被认为不可逾越的障碍。在我的课题中,我提出了一种叫作光激活定位显微镜 (Photoactivated Localization Microscopy, PALM) 的方法,这种方法使得我们能够在发光 (荧光) 分子的帮助下打破限制。利用 PALM 和其它基于发光分子的方法,科学家们深入了解活细胞和单个分子,并大大加深我们对生命的理解。
埃里克 • 白茨格 (Eric Betzig) 教授因在超高分辨率荧光显微技术方面的贡献,与斯特凡 • 赫尔 (Stefan Hell) 教授和威廉 • 默尔纳 (William Moerner) 教授共同获得 2014 年度诺贝尔化学奖。
如何对微小物体观测?
我们在观察物体时,实际上是人眼检测到了从物体反射回来并到达眼睛的光。试想一艘配有声纳探测技术的潜艇,它在大海中航行时,向外发射声波,同时探测水下物体 (如石头和海底生物等) 反射回的声波 (图 1A)。 这样,潜艇中的船员就知道如何在水下航行了。而当我们在实验室观察细胞或微组织时,这一原理也同样适用。我们将光(或其他 辐射 形式如电子等) 照射到要观察的微小物体,观察反射的辐射波,其中,辐射类型由波长决定。或许你知道,波在波峰和波谷间循环,波长 即为两个波峰之间的距离 (图 1B)。
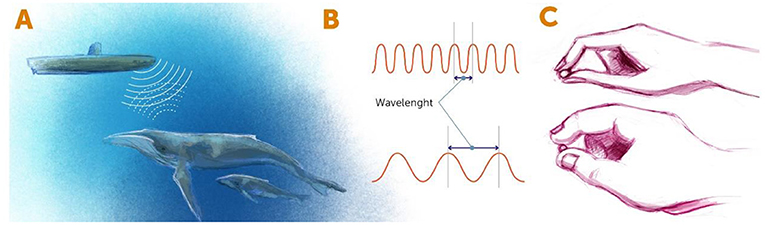
- 图 1 - (A) 利用辐射探测物体。
- 在潜艇的声纳系统中,声波辐射用以探测附近物体。潜艇发射声波,接收到声波辐射的附近物体将其反弹回声纳系统的探测器。同理,人眼 (探测器) 看见物体实际上是接收到了由物体反射回的光波。 (B) 辐射类型可以由波长描述,即两个辐射波相邻波峰之间的距离。 (C) 短波 (上) 类似利用纤细手指感知物体的细节,而长波 (下) 类似胖乎乎的手指只能 “ 看见” 明显的特征。
观察微小物体时,反弹回的能量波长决定了我们会以多高的分辨率观察到它,波长越短,我们能看清的物体就越小。想象一下,当我们用手指去感受一个物体时,手指越厚 (即波长越长),感受物体的细节就越难——就像用圆乎乎的手指辨别毫米级别的特征(图 1C)。所以,如果想要看到物体的细节,有两条路可走。首先,可以选择波长更短的辐射波,如X 射线,但问题是波长短意味着高能量,在高能量辐射下,生命会失去活性,因此对活细胞或生物组织的研究,不能用太短波长的波。另一种方法是使用能量更低的长波,同时设法超越光学衍射极限,看到正常情况下看不到的细节,这就是 超分辨显微成像技术 的核心思想。
在超分辨显微成像技术问世之前,我们只能看到 200 纳米 (即 0.0002 毫米) 以上的活体生物或组织,如动物细胞中较大的细胞器和单细胞生物如细菌等。对于像病毒这样的小生物体或细胞中较小的部分,如单个蛋白质或其他小分子 (图 2A,B),我们无能为力。高分辨率的活体观察是人类历史上的巨大飞跃,这项技术开辟了一个全新的研究领域,具有更好地了解生命最基本过程的发展潜力。传统显微技术无法观测到的细节就这样展露在我们面前,这对生命奥秘的探索来说是一个巨大的惊喜 (图 3A,B)。
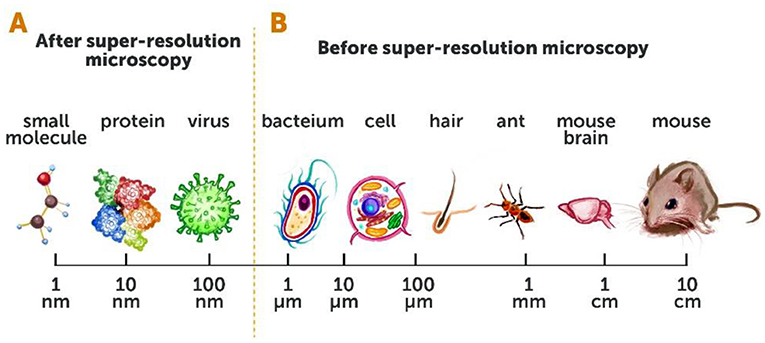
- 图 2 - (A) 传统显微成像技术 vs. 超分辨显微成像技术。
- 超分辨显微成像技术拓展了我们所能看清物体的尺寸范围,小至 10nm(1nm = 0.000000001m) 左右。 (B) 在超分辨显微成像技术问世前,我们只能看见 200nm(大约为一个细菌的尺寸)及其以上大小的物体。
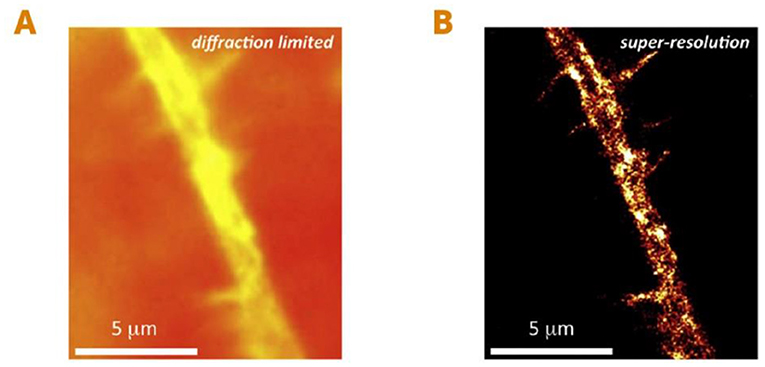
- 图 3 - (A) 利用超分辨显微成像技术对活细胞内的单个蛋白质进行观测。
- 传统显微成像技术下一个活的神经细胞的分支 (B) 超分辨显微成像技术下的同一个神经细胞位置。这种方法帮助我们看清了以前所看不到的细节,在这种情况下,我们看到了神经细胞中负责导电的细胞膜上微小的离子通道蛋白 (黄色的亮点) [1]。
序幕:近场显微技术
1983 年,我在大学进行前沿性研究,两位教授 Mike Isaacson 和 Aaron Lewis 萌生了打破 阿贝衍射极限的疯狂想法。阿贝衍射极限可以解释为:当我们利用光波观察物体时,能看到的物体尺寸最小为光波波长的一半,比如光波波长为 1mm,我们能看到的物体最小为 0.5mm。 教授们决定另辟蹊径,打破此种限制,他们基于 1972 年首次提出的有关突破阿贝衍射极限的理论 [2] 提出了一个设想,其基本思路是在一个黑色小圆盘上钻一个比光波波长小得多的小洞。当黑色圆盘放在物体很近的地方时,一束光穿过小洞,这个物体只有一个很小的点会被照射到,并且这个点的尺寸远远小于光的波长。接下来,通过移动圆盘,就可以实现对物体的 “ 点扫描” 。这样,我们就可以以更高的分辨率观测物体,这种方法即今天的近场扫描光学显微技术 [3, 4]。 这是我研究的第一项超分辨显微成像技术,这种方法的主要问题在于,光通过小孔后发散角很大,为提高分辨率,需要在靠物体很近的地方成像。在对细胞成像时,由于细胞并不是扁平的,很难控制圆盘的位置。我在这个领域深耕几年后,决定退出这个课题,甚至完全放弃了科研。没想到,几年后,生物化学领域的一个巨大突破吸引我重新回到科学和显微成像领域。
超分辨荧光显微成像技术
1994 年,一项前沿性研究横空出世 [5],表明在基因工程的帮助下,我们可以在活细胞的任何蛋白质上连接一个荧光蛋白。 荧光蛋白类似一个发光的把手或 “ 标记物” 。 这种特殊的蛋白质,在特定波长的光的照射下会发光。我迅速意识到,这项工作将在显微成像领域掀起翻天覆地的变化,帮助我们看到细胞内部的微小结构。一年后,即 1995 年,我发表了一篇论文,为革新显微成像技术奠定了基础 [6]。 然而,直到21世纪初,荧光分子领域的进展让我进一步坚定了自己的想法。研究者们开发出了一种荧光分子,可以在特定波长的光照射下得到 “ 激活” 而发光 [7],这意味着我们可以将发光标记物附着在活细胞内部的特定蛋白上,通过有序激活它们而研究细胞结构和运动过程。 光激活定位显微技术 (PALM)[8, 9] 是我协作开发超分辨显微成像技术的起点。2014 年,我因这项技术获得了诺贝尔化学奖。 PALM 的原理是这样的:每个细胞中有 20000 种不同种类的蛋白质,每种蛋白通常有数千个,我们希望了解它们之间的相互作用。在学校的生物课上,在传统显微镜下观察细胞内部的蛋白质时,我们只能看到一个大的发光团。这些蛋白质离得很近,在视野中无法分辨。在PALM 中,我们在蛋白质上附着特殊的荧光标记物,这些标记物在低功率 (紫外光) 激光照射下被激活 (图 4,步骤一),然后经高功率的 (蓝光) 独立激光照射后,它们开始发光并可被探测到 (图 4,步骤二)。
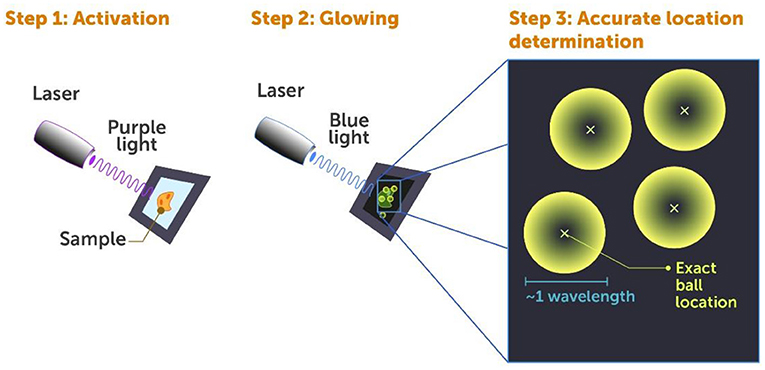
- 图 4 - 光激活定位显微成像技术 (Photoactivated Localization Microscopy, PALM)。
- PALM 中,对荧光标记的细胞进行超分辨荧光成像主要包含三个步骤。第一步:使用能量低的紫外短脉冲光对细胞进行照射,对一部分蛋白上的荧光标记物进行激活准备下一步发光;第二步:蓝色激光照射细胞使其发光并被探测到;第三步:在计算机上通过找到每个发光圆球的中心以确定单个蛋白质的位置。
如果同时激活所有的标记物,它们将会一起发光造成视觉上的混淆,这也是上述我们看到大发光团的原因。因此我们使用能量非常低的紫外脉冲光对标记物进行激活,如此,每个脉冲一次只能随机激活一小部分标记物,并且这些细胞内部被激活的标记物就可以轻松分辨开来。当我们用蓝光对这些激活的标记物进行探测时,它们就像一个个发光的小球 (图 4,步骤二),大小大约为光的一倍波长,这是我们用传统显微镜观测时所能看到的最小尺寸 (图 4,步骤三)。
接下来是此技术的核心步骤。我们使用计算机对采集的图像进行处理,精确找到每个发光小球的中心。你可以想象一个有一定直径的球形篮球,和估计其直径相比,即使你并不能直接看到其中心,也能更为准确地指出球心。同样地,对于这些分子小球也是如此,我们可以以极高的精度找到它们的中心,这比直接确定发光小球的尺寸更接近分子的实际尺寸。这意味着每次我们用光脉冲照射细胞时,都能找到细胞内一小组蛋白质的位置 (图 4,步骤 3)。这些蛋白质的荧光会自然关掉,之后再照射另一组蛋白质并确定其位置。通常,完成对一整个细胞的成像需要激活数万次,但这样的付出是值得的,因为我们获得了一张分辨率极高的细胞图像或是其他研究中的物体图像。(图 3B及 Betzig 等人论文中的图片 [9])。
超分辨显微成像技术的挑战和机遇
如你所知,PALM 十分简单,所需的只是一台产生照明光束的激光器、一台相机和一个用于找到发光蛋白质中心的简易程序。这一套设备既便宜又简单。实际上,我和我的朋友 Harald Hess 教授是在他的客厅里搭建出了第一套 PALM 模型,当时我们都处于失业状态,设备都是自掏腰包。
困难的部分在于对生物样本的处理。我们遭遇了许多棘手的问题,包括生物样品的制备、避免光对细胞的损伤,以及找到一种最佳方法来探测分析我们感兴趣的分子发出的光。
在准备细胞时,实验表明,许多可以激活的标记物并没有附着在我们实际想看的蛋白质上,而是分布在附近的其他物体上。这意味着我们使用的标记物实际上并没有指向我们感兴趣的蛋白质的位置。此外,即使我们设法标记到正确的蛋白质,也只有一小部分被标记,常常不足以以最高的分辨率获得细胞的完整图像。尽管增强光照强度可以获取的信息更多,但如果我们尽力正确标记足够多的蛋白质,细胞可能无法承受如此强烈的光。因此,我们总是在试图获取尽可能多的细胞信息的同时,尽量减少对细胞的损伤。
我要提到的最后一个困扰是光漂白现象。光漂白描述了一个标记物只能发光有限次数的现象。换言之,对于一个特定标记物,在其被彻底破坏前只能发出一定量的光。在某些情况下,这些光不足以让我们提取到足够的信息找到标记物的精确位置。
正如我之前所提及的,超分辨荧光显微技术是一项无与伦比的技术,它让我们得以窥见活细胞和活体组织的奥秘。在这种技术的帮助下,我们不仅能确定活体生物样本的结构,还能在一定时间内跟踪细胞内部的动态过程,如蛋白质的运动。凭借所谓单分子追踪的方法,我们可以深入研究活体细胞的最深处,见证生命的最基本过程 [10, 11]。 例如,这种方法曾经帮助我们探悉 RNA 复制物是如何由细胞核内的 DNA 产生的,这一过程被称为转录。
追踪单个分子并能够探明它们在细胞内的运动,对于新药物的开发至关重要。在我看来,我们以前无法看到但现在可以捕捉到的与细胞机制有关的信息,可能会引领我们迈向一个全新的药物研发阶段,开创针对多种疾病的重要新型治疗方式,如阿尔兹海默症和帕金森症等。我相信,这也许是超分辨显微成像技术带来的最宝贵的贡献,也是我和同事们将我们创办的制药公司命名为 Eikon Therapeutics 的原因。
对年轻人的建议
正如我开头所说,虚构的英雄角色和宇航员激励过童年时的我。在改善人们的生活这件事上,他们厥功甚伟。于我而言,这是一个人在生活中能选择的崇高追求。因此我建议,无论你从事何种职业,去做一些真正有影响力、并能做出有意义贡献的事情。这并不一定是多么伟大的事情,无论是抚养孩子还是打包杂货,都是有价值的。尝试去寻找一些符合你个人兴趣、并能对周围人或社会产生积极影响的事务 (图 5)。如果你真的渴望成为一名科学家,不要深陷于要成为教授的念头中,这并不应该是你的目标,因为除了在大学工作外,还有许多其他可以施展才华的途径。

- 图 5 - 对年轻人的一些建议。
- 当你规划你的未来蓝图时,尝试去做一些你热爱并且对社会有用的事情。
就我个人而言,我发现我做这种研究时有很多优势。首先,我是自己的老板,我享受这种可以自己决策而不是被安排去做事情的感觉;其次,在我的科研领域中,我力图为认真回答我专业范围外科学问题的人们发明新的工具,这意味着我必须学习许多新东西,成为 “ 全能专家” 。我对许多领域的知识都略知一二,从不同的设备中哪种材料表现最好,到生物学和物理学,再到设计新的研究工具,这些广博的知识渗透入我的日常生活中,使得我博物洽闻,畅享这个美妙而复杂的世界。
最后,我想探讨的是应该以什么态度面对自己做出的选择。首先,永远以批判性思维面对所遇到的麻烦,不要满足于表面的机械化思考,要推究根源,寻其本质;第二,不要惧怕冒险,在我看来,这个世界已变得过于安逸,限制了个人和社会创新以及进步的能力;最后,努力工作至关重要!无论你在做什么,处于何种年龄阶段,尝试在快乐中进步,即使前路崎岖,也不要妄自菲薄,要全力以赴去实现目标。如果你对某事并不擅长也没有关系,因为每个人都有不同的专长,没有人是无所不能的。但在做任何事情时都要不遗余力,有志者事竟成。找到你真正热爱的事情,全心全意投入其中,学有所长,尽你所能并乐在其中。
术语表
辐射 (Radiation): ↑ 以波或粒子的形式从某个源头发出的能量。
波长 (Wavelength): ↑ 波的一个参数,具体为两个相邻波峰之间距离。
超分辨显微成像技术 (Super-resolution microscopy): ↑ 任何一种可突破衍射极限并帮助我们以更高分辨率看清物体的显微成像技术。
阿贝衍射极限 (Abbe’s diffraction limit): ↑ 光学显微镜的物理极限,意为当两个点之间的距离小于可见光波长的一半时,我们将无法分辨它们。
近场扫描光学显微成像技术 (Near-Field Scanning Optical Microscopy): ↑ 20 世纪 80 年代提出的第一种超分辨显微成像技术。
光激活定位显微成像技术 (Photoactivated localization microscopy (PALM)): ↑ 我发明的一种利用荧光分子打破阿贝衍射极限的超分辨显微成像技术。
光漂白 (Photobleaching): ↑ 荧光材料发光一定次数后失去发光能力的一种现象。
利益冲突声明
作者声明, 该研究是在没有任何可能被解释为潜在利益冲突的商业或财务关系的情况下进行的。
致谢
感谢 Noa Segev 的采访和对文章撰写的参与,以及Alex Bernstein 提供的数据。
感谢 “ 赛先生” 公众号及其译者杨晓雨、任知微、李栋对本文中文翻译的贡献。
扩展阅读
- Eric Betzig and Harald Hess (Janelia Farm/HHMI): Developing PALM Microscopy.
- Prof. Betzig Nobel Lecture.
参考文献
[1] ↑ Ondrus, A. E., Hsiao-lu, D. L., Iwanaga, S., Parsons, W. H., Andresen, B. M., Moerner, W. E., et al. 2012. Fluorescent saxitoxins for live cell imaging of single voltage-gated sodium ion channels beyond the optical diffraction limit. Chem. Biol. 19:902–12. doi: 10.1016/j.chembiol.2012.05.021
[2] ↑ Ash, E. A., and Nicholls, G. 1972. Super-resolution aperture scanning microscope. Nature 237:510–2. doi: 10.1038/237510a0
[3] ↑ Betzig, E., Harootunian, A., Lewis, A., and Isaacson, M. 1986. Near-field diffraction by a slit: implications for superresolution microscopy. Appl. Opt. 25:1890–900. doi: 10.1364/AO.25.001890
[4] ↑ Betzig, E., and Chichester, R. J. 1993. Single molecules observed by near-field scanning optical microscopy. Science 262:1422–5. doi: 10.1126/science.262.5138.1422
[5] ↑ Chalfie, M., Tu, Y., Euskirchen, G., Ward, W. W., and Prasher, D. C. 1994. Green fluorescent protein as a marker for gene expression. Science 263:802–5. doi: 10.1126/science.8303295
[6] ↑ Betzig, E. 1995. Proposed method for molecular optical imaging. Opt. Lett. 20:237–9. doi: 10.1364/OL.20.000237
[7] ↑ Patterson, G., and Lippincott-Schwartz, J. 2002. A photoactivatable GFP for selective photolabeling of proteins and cells. Science 297:1873–1877. doi: 10.1126/science.1074952
[8] ↑ Shroff, H., White, H., and Betzig, E. 2013. Photoactivated localization microscopy (PALM) of adhesion complexes. Curr. Protocol. Cell Biol. 58:4–21. doi: 10.1002/0471143030.cb0421s58
[9] ↑ Betzig, E., Patterson, G. H., Sougrat, R., Lindwasser, O. W., Olenych, S., Bonifacino, J. S., et al. 2006. Imaging intracellular fluorescent proteins at nanometer resolution. Science 313:1642–5. doi: 10.1126/science.1127344
[10] ↑ Liu, Z., Lavis, L. D., and Betzig, E. 2015. Imaging live-cell dynamics and structure at the single-molecule level. Mol. Cell. 58:644–59. doi: 10.1016/j.molcel.2015.02.033
[11] ↑ Li, D., Shao, L., Chen, B. C., Zhang, X., Zhang, M., Moses, B., et al. 2015. Extended-resolution structured illumination imaging of endocytic and cytoskeletal dynamics. Science 349:aab3500. doi: 10.1126/science.aab3500