
Abstract
Cystic fibrosis (CF) is a serious genetic disease that causes thick, sticky mucus to build up in the lungs and digestive system. For many years, treatments could only manage the symptoms. That began to change with the discovery that CF is caused by problems with a protein called CFTR, which normally helps keep mucus thin and slippery. When CFTR is missing or does not work properly, mucus becomes too thick, leading to serious health problems. After scientists discovered how CFTR works and what goes wrong in CF, another team began developing medicines that help the faulty protein fold correctly, reach the cell surface, and open to let chloride pass through. Today, powerful triple therapies that combine these drugs work for about 90% of people with CF. This work has changed what it means to live with the disease—and has shown how understanding the root of a problem can lead to real solutions.
Drs. Welsh and Negulescu were awarded the 2025 Gairdner International Award “For pioneering research into the cellular and molecular mechanisms underlying the genetic disease cystic fibrosis, leading to the development of transformative drug therapies based on these mechanisms, thereby improving and saving countless lives”.
What is Cystic Fibrosis?
Your body makes mucus every day. Sometimes this is obvious—like when you have a runny nose or a wet cough. But even when you feel fine, mucus is constantly produced in places like your lungs, nose, and digestive system. This thin, slippery mucus does some important jobs: it keeps your airways moist, traps dust and germs, and helps move food through the intestines. However, in people with a condition called cystic fibrosis (CF), the mucus becomes thick and sticky. Instead of flowing smoothly, it clogs the lungs and digestive system, making it harder to breathe, harder to digest food, and easier for infections to grow.
CF is a genetic disease, which means people are born with a harmful change in one of their genes. It affects more than 100,000 people worldwide, including over 40,000 in North America. Most people with CF are diagnosed as babies or young children. For many years, doctors could only treat the symptoms of CF: clearing mucus from the lungs, helping with digestion, and treating infections. These treatments helped, but they did not stop the disease from getting worse. Not long ago, many people with CF did not live past their teens or twenties.
My name is Michael Welsh, and I first became interested in cystic fibrosis during medical school. I still remember one of the first young patients I met—a girl around 7 or 8 years old. Even before I opened the exam room door, I could hear her coughing. When I walked in, I saw how hard she had to work just to breathe. I smelled a grape-like smell and later learned that it was due to her lung infection with bacteria called Pseudomonas aeruginosa. Talking with her and her parents, I saw how much the disease had taken from her. That moment never left me, and it made me want to understand what was really going wrong in the bodies of people with CF. What exactly was broken—and could it be fixed?
A Clue Hidden in the Cells
Back in 1989, scientists discovered the gene that causes CF. They named it cystic fibrosis transmembrane conductance regulator (CFTR). When the gene was first found, no one knew what the CFTR protein actually did inside the body, or how a broken version of it could cause such serious symptoms.
My team and I wanted to understand CFTR’s job. We discovered that CFTR is a type of protein called an ion channel—a tiny tunnel that sits in the membrane that covers the surface of cells where it helps chloride, a type of salt, flow in and out across the cell membrane (Figure 1A) [1]. Where chloride moves, water follows, so chloride movement helps control how much water stays in the thin layer of mucus that coats and protects the lungs and digestive system. When CFTR is working, chloride flows out of cells, and so does water. The water keeps the mucus thin and slippery, so it can do its job. If CFTR is missing or does not work properly—often because of harmful changes called mutations in the CFTR gene—chloride cannot flow, and water cannot follow. In people with CF, lack of water makes the mucus thick and sticky.
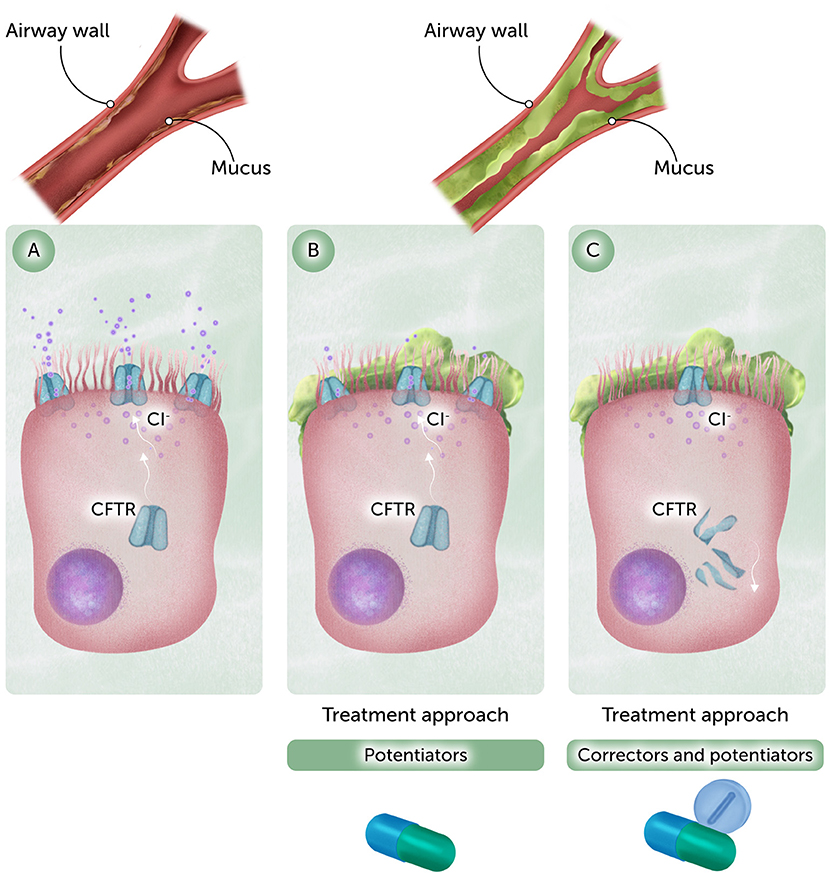
- Figure 1 - (A) Normal CFTR allows chloride to flow across the cell membrane, keeping mucus thin and slippery.
- (B) With gating mutations, CFTR is present on the cell surface, but it does not let chloride through properly. Drugs called potentiators can improve CFTR function. (C) In the most common mutation, ΔF508, a reduced amount of CFTR is found on the cell surface because it does not fold properly and is broken down inside the cell. The CFTR that makes it to the surface does not function properly. A combination of potentiators and correctors (which help CFTR reach the cell surface) can treat this condition (Figure credit: Somersault18:24).
Figuring this out was not easy and it took years. We had to use whatever tools we could find, even ones invented for completely different purposes. One method we used to study chloride flow came from frog skin research. Other techniques were originally developed for studying nerve cells or viruses. We adapted all of them to study CFTR. That kind of flexibility—using the best tools available, no matter where they came from—made a big difference. Understanding how CFTR worked gave us a solid starting point. For the first time, we could begin to look for ways to fix the root of the problem.
What Makes the Channel Work?
After we discovered that CFTR is a chloride channel, we wanted to understand how the tunnel opens and closes. What tells it when to let chloride flow through? We found that CFTR needs a kind of chemical tag called a phosphate group to get ready to open. Only when that tag is in place can the protein respond to signals that tell it to open.
While phosphate tagging gives a CFTR channel “permission” to open, another molecule called ATP provides the switch that causes CFTR to open and close. When ATP binds to CFTR the channel changes its shape, allowing chloride to flow through. Understanding what CFTR needs to function gave us another important clue. It helped explain how certain mutations might cause trouble—and pointed to new ways scientists might be able to help the protein work better.
Not All Mutations are the Same
Our discoveries up to this point helped explain what CFTR does and how it works. But how do mutations disrupt its normal function? The answer turned out to be complicated. Scientists found not just one or two mutations in the CFTR genes of people with CF—there are hundreds. Our lab and others discovered that there are several ways mutations can affect the CFTR protein and prevent it from working properly [2].
In some cases, the CFTR channel does not open when it should, or it might not stay open long enough to let enough chloride flow through (Figure 1B). Some mutations prevent CFTR from folding into its normal shape. As a result, after it is made, the cell’s quality-control system recognizes CFTR as defective and breaks it down, so it never reaches the cell surface.
One mutation, called ΔF508, is the most common. It causes problems in two ways: it affects how the protein folds and how the channel opens (Figure 1C). At first, many people thought this kind of damage would be too hard to fix. But we found early clues that the protein might still work if given the right support. That gave us hope.
As we studied more mutations, we began to sort them into categories based on how they interfered with CFTR. This helped scientists understand that no single treatment would work for everyone. But it also gave us something hopeful: the idea that we might be able to help more people by matching treatments to the specific problems caused by their mutations.
Fixing The Protein—A New Kind of Medicine
My name is Paul Negulescu, and I led a team at Vertex Pharmaceuticals that set out to take what Michael and others had learned about CFTR and turn it into medicines. Our goal was not just to treat the symptoms of CF, but to help the faulty protein itself work better.
We started by searching for small drug-like compounds that could help fix the ΔF508-CFTR protein. To do that, we developed specialized lab tests that could measure how well CFTR was working in cells [3]. Then we tested hundreds of thousands of compounds, one by one, using robotic systems. Most did not show improvements, but a few did. Over time, we made better versions of the ones that seemed to help, and we tested thousands of the best ones in human CF airway cells in the lab (Figure 2) [4]. At the time, most research groups tested the effectiveness of new drugs in animals like mice. But we believed that testing compounds on human cells would give us a better chance to select compounds that would work well in people. Michael’s team showed us how they grew the airway cells and measured CFTR in those cells, and then we built a system to test compounds.
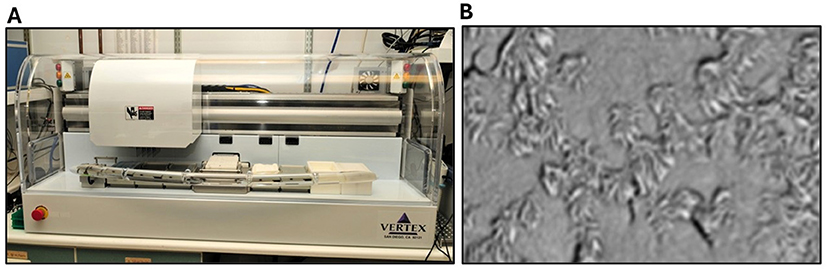
- Figure 2 - (A) The robotic platform that we used to test thousands of compounds in human airway cells, to see if they could improve the way faulty CFTR proteins worked.
- (B) A microscope view of human airway cells used to test the activity of potentiators and correctors. We tested the compounds on human cells since CF is a human disease. The hair-like structures on the outside of the cells are cilia, which act like little brushes to clear mucus and debris from the lungs.
Over time, we identified two types of compounds that made CFTR work better. The first type we called potentiators. These compounds helped the CFTR protein open more easily. After several years of work, we developed one potentiator, called Ivacaftor, that worked well. By itself, it improved the function of CFTR with specific gating mutations—mutations that affect how CFTR opens and closes. Ivacaftor was the first drug to treat the underlying cause of cystic fibrosis, not just the symptoms. Finding correctors, which help correct CFTR processing and allow it to reach the cell surface, took almost a decade longer! But eventually we were able to develop both potentiators and correctors—each solving a different part of the problem.
A Triple Therapy that Changed Everything
Ivacaftor was a big step forward, but it only worked for a small group of people with certain gating mutations. Most people with CF, including those with the most common ΔF508 mutation, still needed a corrector. That is because ΔF508 causes two kinds of trouble: the CFTR protein folds incorrectly and does not open properly, even if it gets to the cell surface.
To solve that, we worked on combining drugs [4]. Eventually, we developed a three-drug combination that contained two correctors (to help CFTR reach the cell surface) and one potentiator (to help it work once it got there). This triple therapy is now known as Trikafta, and it works for about 90% of people with CF! In studies with CF patients, Trikafta improved lung function, reduced hospital visits, and helped people gain weight and feel stronger. A new triple therapy, called Alyftrek, works similarly and may further improve CFTR function.
Seeing how much better people felt on Trikafta was incredibly rewarding. For many patients, it changed CF from a life-shortening disease into a condition they can manage—something they live with, not die from. But the work of both my team and Michael’s was also a reminder of how much science had to happen first, and how long it took. Our work to discover and develop the medicines was based on efforts by a much larger “team”: the doctors who first noticed patterns in patients decades ago, researchers who spent years studying chloride and cell biology, and people with cystic fibrosis who volunteered for studies. I am also very grateful for the inspiration and support of the Cystic Fibrosis Foundation to begin this project. Everyone played a part in making these breakthroughs possible. It was a true bench-to-bedside effort—from basic science in the lab to medical treatments that changed people’s lives.
Changing What is Possible
Because of our work, many people with CF are now living longer, healthier lives. Treatments that target the root cause of the disease have changed what is possible—for patients, families, and doctors. But the impact goes even further. Our discoveries helped show that it is possible to repair a faulty protein, even when the problem appears complicated. That idea is now inspiring new research into other diseases caused by gene mutations.
This journey also reminded us that scientific progress takes time, teamwork, and determination. It means using every tool you can, learning from setbacks, and believing that even the hardest problems might have a solution. And it means listening to the people you are trying to help. One patient once told me she joined a research study even though it would not help her, because it might help someone else. This made me feel like failure was not an option—not when people were counting on us. Stories like this help us remember that our work is not finished, because about 10% of people with CF still do not respond to the current therapies.
To anyone interested in science or medicine, our advice is simple: be curious. Ask questions. Try new things. Do not worry about what others expect—focus on what matters to you. If you throw yourself into something fully, you will figure out whether it is the right path. And when you work as part of a team, remember that your effort can help someone else do their best work, too. That is what makes breakthroughs possible.
Glossary
Cystic Fibrosis: ↑ A genetic disease that causes thick, sticky mucus to build up in the lungs and digestive system, making it harder to breathe, digest food, and fight infections.
Ion Channel: ↑ A tunnel-like protein in the cell membrane that lets charged particles (like chloride and other salts) move in or out of the cell, helping control fluid balance, nerve signals, and more.
Mutation: ↑ A change in a gene’s DNA sequence. Some mutations can cause diseases by making a protein that does not work properly or not making the protein at all.
ATP: ↑ A molecule that stores and provides energy for many processes inside cells—kind of like a battery that controls cellular machines, including some proteins like CFTR.
Potentiators: ↑ Medicines that help a protein channel open more easily or stay open longer, allowing it to work better. Used to treat some forms of cystic fibrosis.
Gating Mutation: ↑ A type of mutation that affects how a protein channel opens and closes. In cystic fibrosis, gating mutations can keep CFTR from opening properly to let chloride through.
Correctors: ↑ Medicines that help a faulty protein fold the right way so it can reach the surface of the cell and do its job—used to treat cystic fibrosis.
Conflict of Interest
PN was employed by the company Vertex Pharmaceuticals.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We wish to thank Dr. Susan Debad for her thoughtful questions, collaborative input, and her contributions as co-author. Figure 1 was created by Somersault18:24.
AI Tool Statement
The author(s) declare that no Gen AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
References
[1] ↑ Anderson, M. P., Gregory, R. J., Thompson, S., Souza, D. W., Paul, S., Mulligan, R. C., et al. 1991. Demonstration that CFTR is a chloride channel by alteration of its anion selectivity. Science 253:202–5.
[2] ↑ Welsh, M. J, and Smith, A. E. 1993. Molecular mechanisms of CFTR chloride channel dysfunction in cystic fibrosis. Cell 73:1251–4.
[3] ↑ González, J. E., Oades, K., Leychkis, Y., Harootunian, A., and Negulescu, P. A. 1999. Cell-based assays and instrumentation for screening ion-channel targets. Drug Discov. Today 4:431–9. doi: 10.1016/S1359-6446(99)01383-5
[4] ↑ Van Goor, F., Straley, K. S., Cao, D., González, J., Hadida, S., Hazlewood, A., et al. 2006. Rescue of ΔF508-CFTR trafficking and gating in human cystic fibrosis airway primary cultures by small molecules. Am. J. Physiol.-Lung Cell. Mol. Physiol. 290:L1117–30. doi: 10.1152/ajplung.00169.2005