
Abstract
Lysosomes are specialized cell structures that serve as the garbage collection system of the body‘s cells. The lysosome contains components that clean up various types of cellular “trash” and are responsible for activating the recycling process. When lysosomes do not work properly, trash accumulates in the cells, eventually leading to a sick cell and cell death. This can lead to the development of several lysosomal storage diseases. Symptoms of these diseases vary in severity, from symptoms that barely affect a patient’s life up to symptoms already starting at birth that greatly reduce life expectancy. Two treatment options are available for some of these diseases that can ameliorate symptoms, the enzyme replacement therapy and substrate reduction therapy. A new research approach called gene therapy, provides a potential cure. In this article, we will explain the role of lysosomes and what happens when they do not work properly. We will also provide details about the available treatments and how gene therapy could be a breakthrough in the field.
What are Lysosomes?
Lysosomes are specialized structures within cells, each surrounded by a membrane (displayed in Figures 1c, d as cut view). They contain many types of enzymes, (displayed in detail in Figure 1c), which are specialized proteins needed to fulfill the lysosomes’ duties. Lysosomes can be found in nearly all types of animal cells, including human cells. There are 50–1,000 lysosomes in each cell. When researchers started to analyze the function of lysosomes, they thought lysosomes were only the garbage-collection system of cells. They observed that lysosomes take up all cell garbage and, with the help of enzymes, digest it like a recycling center that shreds garbage before using it to make new materials (Figures 1a, c). Now it is known that the function of lysosomes is much more complex. They also provide garbage to other recycling centers and are involved in the repair of damaged cell membranes. Even more importantly, lysosomes function as sensor (Figures 1a, c) that can tell if a cell is healthy, if it is missing nutrients, or if it has been attacked by bacteria or viruses. Once the lysosome senses such a problem, it can take appropriate measures to improve the cell’s wellbeing (displayed as antenna sensing smell in Figures 1a, c). It can, for example, inform the immune system about intruders, so that the body’s defenses can become active and fight against dangerous invaders. Therefore, lysosomes are also important watchmen, keeping cells healthy [1].
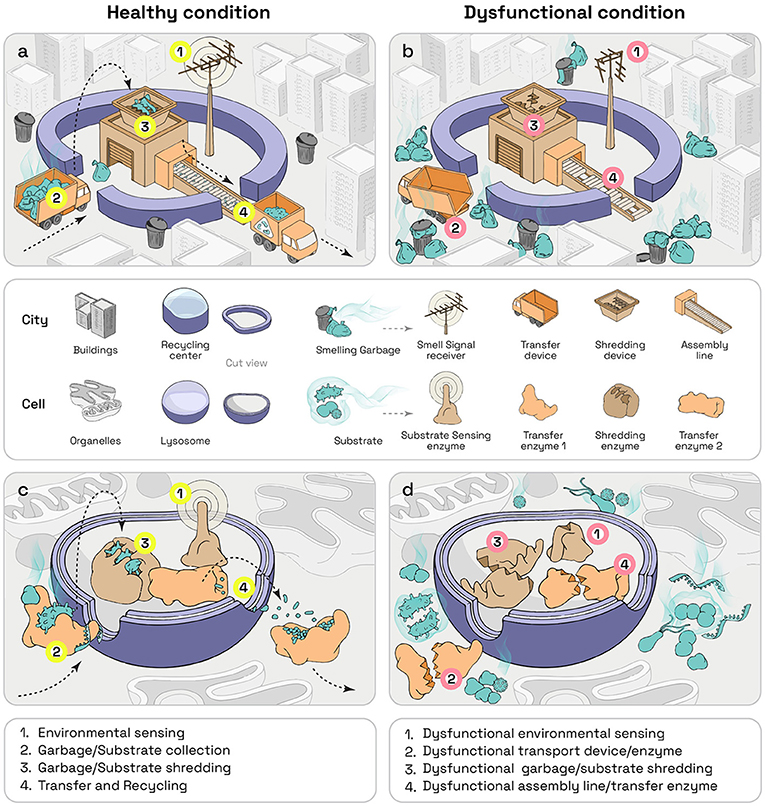
- Figure 1 - The lysosome and its functions.
- Healthy (a) and dysfunctional (b) lysosome presented as garbage collector and recycling center. (c, d) Lysosomal functions on cellular level. (a, c) In the healthy situation, the lysosome can sense the environment of the cell (1). It further collects trash (2), breaks it down (3), and recycles it (4). In lysosomal storage diseases [dysfunctional condition, (b, d)], one of these functions is not working. For example, the sensor (1), the collector (2) the shredding system (3) or the recycling system (4), can be broken. In these cases, cellular trash builds up until cells get sick.
Lysosomal Storage Diseases
What happens when lysosomes do not work as they are supposed to? Disturbances of the lysosomal system can have many causes, as more than 60 enzymes are involved in proper lysosomal function. In a lysosomal storage disease, one of the enzymes, or a protein that is important for lysosome function does not work properly. This defect causes an accumulation of trash that the enzyme was supposed to recycle (Figures 1b, d). Just imagine the garbage collectors of your neighborhood removing all the garbage that you sorted and placed outside for collection, except for the plastic trash. After a few weeks, streets would fill up with plastic trash. That is exactly what happens in diseased cells—cellular garbage that can no longer be recycled clogs the cell and eventually kills it. Scientists call this garbage substrate (Figures 1b, d). Since each of the enzymes handles a separate task, the cell—and consequently the patient —experiences specific problems depending on which enzyme does not function properly. By now, more than 60 lysosomal storage diseases are known, which can affect various parts of the body, like the skeleton, brain, skin, or heart, depending on the affected enzyme.
Lysosomal storage diseases belong to the group of rare diseases. A disease is defined to be rare if it does not affect more than 1 out of 1,500 people. Taken together, all the lysosomal storage diseases affect about 1 out of 4,000 people. Due to their infrequency, doctors rarely see lysosomal storage disease patients, so they do not have a lot of experience with them. This means that patients with lysosomal storage diseases might struggle for years to get a correct diagnosis. Most lysosomal storage diseases are severe and result in an early death. Some children die shortly after birth, while other patients experience the first symptoms during adulthood [2]. Many lysosomal storage diseases are named after the physician(s) who first described them, like Krabbe, Gaucher, Niemann-Pick, Hunter, Hurler, and Tay-Sachs disease. Additionally, all of these diseases are named based on the defective protein, for example mucopolysaccharidosis, mucolipidosis, gangliosidosis, sphingolipidosis, and many more. Diseases named after the physicians thus have two names, the common name based on the physician, plus the name based on the defective protein defect.
The Case of Hannah
Barely two days after Hannah was born, doctors realized that something was wrong, as her spleen was larger than normal. Finding the right diagnosis took doctors 5 months, and Hannah was diagnosed with a severe form of Gaucher disease that also affects the brain. Doctors gave her 9 months to live. During the next months, Hannah’s coordination suffered and her development was strongly delayed. In addition to her spleen, her liver was affected, and she was no longer able to sit unassisted or grab things on her own. Although she seemed to be a happy toddler, her brain was eventually destroyed by the disease. Hannah lived 3 years before she passed away in her parents‘ arms. If you would like to learn more about Hannah, visit this site.
Is There a Treatment or Cure?
As most lysosomal storage diseases have severe symptoms, researchers are urgently searching for a cure. So far, the therapies that are available can only improve symptoms—a full cure is currently not possible. There are a few therapy options that are already used for some lysosomal storage diseases or that are currently under development. This section will describe those options and their challenges.
Enzyme Replacement Therapy
During enzyme replacement therapy, the patient is treated with the enzyme that is not produced by their own cells. As a lysosomal storage disease can be caused by a lack of only one of many lysosomal enzymes, each of these diseases needs its own enzyme replacement drug (Figure 2a). By providing the body with the missing enzyme, the lysosome can work properly, therefore preventing the accumulation of cellular trash, the substrate. Enzyme replacement therapy can keep the cells functional and healthy. For a successful treatment, enzyme replacement therapies must be started as early as possible to avoid heavy build-up of substrate. As enzymes break down quickly in the body, they must be regularly injected for the rest of the patient’s life. Another problem with enzyme injections is that enzymes cannot reach the brain due to the blood-brain barrier, which protects the brain from everything that might potentially harm it. This barrier blocks enzymes from entering the brain as they are too large (Figure 2a). As many lysosomal storage diseases are caused by malfunctioning enzymes that are also important for proper brain function, enzyme replacement therapy has only a limited benefit for such patients [3].
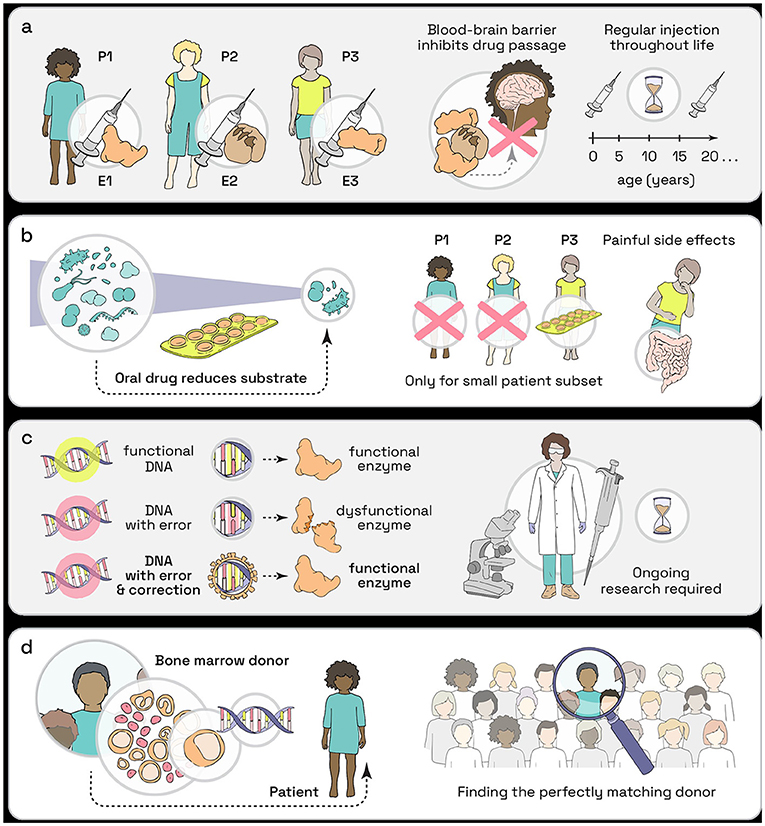
- Figure 2 - Treatment of lysosomal storage diseases.
- (a) Enzyme replacement involves regular injections of the enzyme but it can’t reach the brain. (b) In substrate reduction, patients can take pills, but this comes often with strong side effects. (c) Gene therapy tries to fix the underlying reason of a disease and might be able to provide a cure. (d) Bone marrow donation is already used, but it is difficult to find a perfectly matching donor (P, patient; E, enzyme).
Substrate Reduction Therapy
An alternative approach to improve patients’ life’s is substrate reduction therapy, which uses drugs that reduce the production of the substrate. This means that, instead of cleaning up the trash, substrate reduction therapy minimizes trash build-up. So, if your garbage collection system does not work for plastic trash, produce as little plastic trash as possible! A big advantage of substrate reduction therapy is that it is taken orally as a pill, although it is currently available for only a few lysosomal storage diseases (Figure 2b). Also, big disadvantages of these therapies are the strong side effects that many patients develop, like diarrhea and stomach problems [4].
Gene Therapy
Lysosomal storage diseases are mostly caused by a single defect in the gene of a lysosomal enzyme; it is like an error in the protein‘s construction plan. A new therapy approach aims to fix the underlying gene so the cells can produce the enzyme and recycle trash once again. Even though properly functioning enzymes are needed in the whole body, rescuing the improperly functioning enzyme in some of the patient’s cells might produce enough enzyme to prevent disease symptoms or keep them to a minimum. Treating a disease by fixing a gene, the DNA, is called gene therapy and it would need to be performed individually for every patient, as patients most likely have unique gene defects (Figure 2c). Currently, researchers are searching for the best method to fix a patients’ genes. For example, they try using modified viruses to safely introduce the healthy gene into the patients’ cells. Experiments exploring this therapy are performed in cells that were donated from lysosomal storage disease patients. All experiments are thus performed outside of the body, to make sure that no patients are harmed.
Bone Marrow and Blood Cell Donation
An already established alternative to gene therapy is the treatment of lysosomal storage disease patients with bone marrow or blood cells from a healthy human donor. This is similar to a kidney or liver transplant. The problem with this method is that the donor needs to be a perfect biological match with the patient, which is hard to find (Figure 2d). Although these new and healthy cells often produce enough enzyme to prevent major damage to the organs, it is not enough for the patient to be free of symptoms [5].
Summary
Although lysosomal storage diseases are very rare, their symptoms can be various and severe. Currently, there are no perfect treatment options available, but researchers keep on exploring. Specifically, the gene therapy approach is expected to result in individualized treatment options and maybe even a cure for lysosomal storage disease patients. In the meantime, patients can be helped with a variety of drugs that must be chosen for each patient individually.
Glossary
Lysosome: ↑ Specialized cell structure, surrounded by a membrane, that serves as the garbage collection system of the body‘s cells.
Enzymes: ↑ Specific type of protein that can speed up a chemical reaction in the body.
Lysosomal Storage Disease: ↑ Diseases caused by not properly functioning lysosomes.
Substrate: ↑ Cellular garbage that can no longer be recycled and thus clogs the cell and eventually kills it.
Enzyme Replacement Therapy: ↑ Treatment with an enzyme that restores the function of an enzyme that is missing in the body.
Substrate Reduction Therapy: ↑ Treatment that reduces the production of a substrate.
Gene Therapy: ↑ Fixing a defective gene that otherwise causes a disease.
Donor: ↑ A person who voluntarily provides some of its body’s healthy cells or tissue
Conflict of Interest
SF is employed by BioDoks e.U. and QPS Austria GmbH. TH is employed by Scillustration.
References
[1] ↑ Inpanathan, S., and Botelho, R. J. 2019. The lysosome signaling platform: adapting with the times. Front. Cell Dev. Biol. 7, 113. doi: 10.3389/fcell.2019.00113
[2] ↑ de O. Poswar, F., Vairo, F., Burin, M., Michelin-Tirelli, K., Brusius-Facchin, A. C., Kubaski, F., et al. 2019. Lysosomal diseases: overview on current diagnosis and treatment. Genet. Mol. Biol. 42, 165–177. doi: 10.1590/1678-4685-gmb-2018-0159
[3] ↑ Fernández-Pereira, C., Millán-Tejado, B. S., Gallardo-Gómez, M., Pérez-Márquez, T., Alves-Viller, M., Melcon-Crespo, C., et al. 2021. Therapeutic approaches in lysosomal storage diseases. Biomolecules 11, 1775. doi: 10.3390/biom11121775
[4] ↑ Coutinho, M., Santos, J., Matos, L., and Alves, S. 2016. Genetic substrate reduction therapy: a promising approach for lysosomal storage disorders. Diseases 4, 33. doi: 10.3390/diseases4040033
[5] ↑ Nagree, M. S., Scalia, S., McKillop, W. M., and Medin, J. A. 2019. An update on gene therapy for lysosomal storage disorders. Exper. Opini. Biol. Ther. 19, 655–670. doi: 10.1080/14712598.2019.1607837