
Abstract
When something is rare it means that it happens very infrequently. Did you know that most diseases are rare? There are more than 6,000 known rare diseases, each affecting fewer than 1 in every 2,000 people. But if we put all the rare diseases together, they affect about 1 in 17 of us! Given that they are individually uncommon, rare diseases are often poorly understood. However, rare diseases have a large impact on families and society, thus they require increased attention. In this article, we will explore a rare disease of the nervous system called spinal muscular atrophy (SMA). We will tell you about the symptoms of SMA and explain how it is inherited. SMA has led the way in the discovery of treatments for rare diseases. Finding treatments for rare diseases requires intensive research and commitment from many people, but the success of SMA treatments highlights the importance of studying other rare conditions.
Rare Diseases
Diseases occur when body parts malfunction. For example, Alzheimer’s disease damages the brain, and cardiovascular disease affects the heart. These conditions are some of the leading causes of death and are therefore well-studied by scientific researchers around the world. However, there are many other lesser-known diseases that seriously impact patients and their families, and most of these diseases receive much less attention because they are rare.
There are over 6,000 known rare diseases, including cystic fibrosis, Duchenne muscular dystrophy, and sickle cell disease. Rare diseases affect fewer than 1 in every 2,000 people. Very few of these diseases have cures and most do not even have effective treatments to help patients who suffer from them. However, although each individual rare disease does not affect many people, it is estimated that 1 in every 17 people are affected by some kind of rare disease in their lifetimes. This means that, when taken together, rare diseases have a big impact—not only on a person’s health and wellbeing, but also on their finances, their families, and society. This situation will only improve if rare diseases are carefully studied, both by doctors (to improve diagnosis and understanding of symptoms) and scientists (to identify which faulty molecules cause the disease and to develop treatments).
What Is Spinal Muscular Atrophy?
Spinal muscular atrophy (SMA) is a rare disease of the nervous system that affects ~1 in 10,000 people. People with SMA have muscle weakness and atrophy (wasting away/shrinking) that gets worse over time and affects their mobility [1]. Symptoms of SMA generally begin before 6 months of age. Without help for their breathing difficulties (or one of the new treatments discussed below), most children with SMA do not live beyond their second birthday.
To move our bodies, to swallow, and to breathe, our muscles must contract and relax. Muscle contraction is triggered by electrical signals from the brain, which are sent along nerve cells called motor neurons (Figure 1A). The command centers of these cells, called cell bodies, are found within the spinal cord and have long, thin extensions called axons. Axons connect the cell body to individual muscles, where they form specialized connections that help with nerve-to-muscle communication and muscle contraction (Figure 1B).
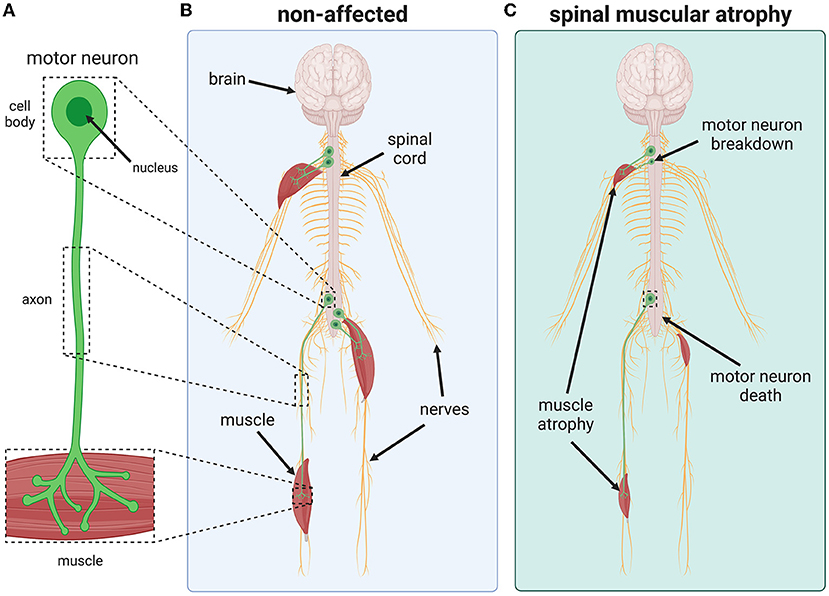
- Figure 1 - SMA is a rare disease that causes motor neurons to die.
- (A, B) Found within the spinal cord, motor neurons are nerve cells that have long, thin extensions called axons that connect to muscles. Healthy motor neurons transmit electrical signals from the spinal cord to the muscle, causing the muscle to contract and the body to move. (C) In people who have SMA, motor neurons break down and die, destroying the contact between the nervous system and the muscles. This results in muscle weakness and atrophy due to reduced use.
In SMA, motor neurons do not work properly and begin to break down and eventually die (Figure 1C). Motor neurons cannot be replaced so, when they die, electrical signals no longer reach the muscles. This prevents the muscles from contracting and, over time, muscles that are not used weaken and atrophy.
What Causes SMA?
SMA, like most rare diseases, is a genetic condition. This means that it is inherited through the DNA, which is passed on from parents to children. Genes, which are short sections of DNA that code for proteins, sometimes become damaged or lost by a process called mutation (for details about mutations, see this Frontiers for Young Minds article). As humans have more than 20,000 genes, we can inherit many rare diseases through mutations.
SMA is caused by mutations in a gene called Survival Motor Neuron 1 (SMN1) [1]. SMN1 normally produces a protein called SMN (Figure 2A). All cells need SMN to stay healthy, but it is particularly important for motor neurons. When the SMN1 gene is mutated, it no longer produces enough SMN protein for motor neurons to survive, so these cells break down and die in SMA patients (Figure 2B).
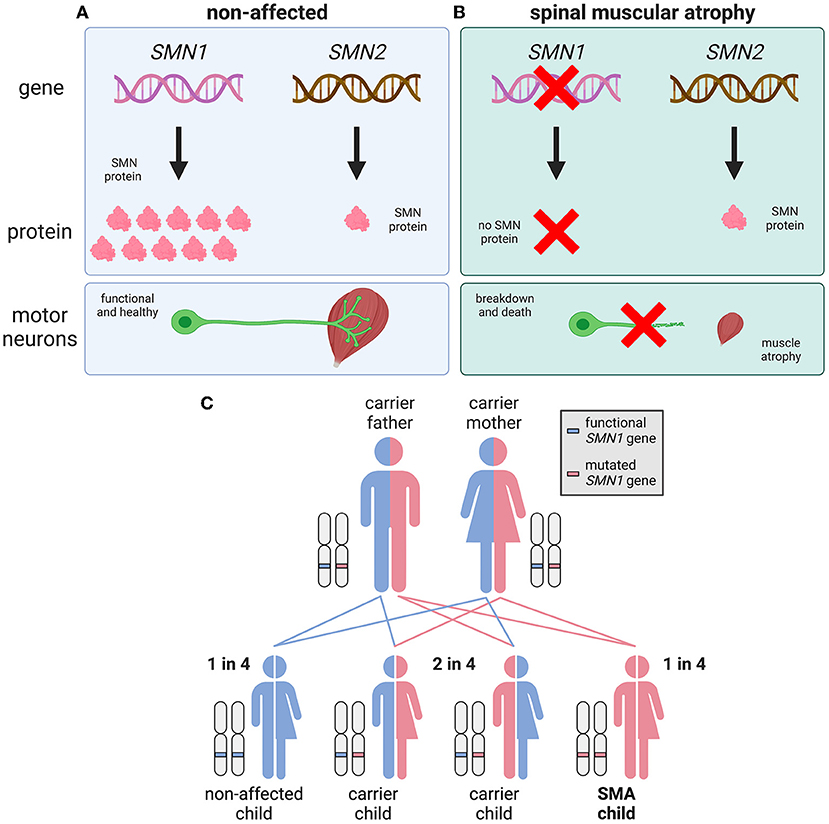
- Figure 2 - SMA is caused by mutations in the SMN1 gene.
- (A) In healthy people, the SMN1 and SMN2 genes produce SMN protein, which is needed by motor neurons to survive. SMN1 produces all the required SMN, while SMN2 makes a much smaller amount. (B) SMN1 is mutated or deleted in people who have SMA, and it no longer makes SMN. A small amount of SMN is made by SMN2, but this is not enough for motor neurons to survive. (C) SMA is an autosomal recessive disease, which means that two faulty/mutated copies of SMN1 must be inherited for SMA to develop.
Unlike most proteins, SMN can also be made by a second gene, named SMN2. However, SMN2 makes 10 times less SMN protein than SMN1. In people who do not have SMA, SMN2 is not needed—SMN1 makes plenty of SMN to keep cells healthy. However, people with SMA must rely only upon SMN2 for their SMN protein, because their SMN1 genes do not work.
You may have heard that people have only two copies of each gene—one inherited from mom and one from dad. However, this is not true for every gene and, luckily, SMN2 is one of those exceptions. Caused by an error, genes can sometimes be accidentally duplicated, which leads to there being more than the usual two copies. For people with SMA, the more copies of SMN2 they have, the more SMN they make—and the less intense their symptoms. This is why some cases of SMA are severe and others are milder. This also means that small increases in SMN could improve the quality of life for a person with SMA. This is important for the treatment options discussed below.
How Is SMA Inherited?
SMA is an autosomal recessive condition, which means that two faulty copies of the SMN1 gene must be inherited (one from each parent) for the disease to occur (Figure 2C). The word “autosomal” refers to the disease gene being found on one of the 22 non-sex chromosomes, known as autosomes. When a condition is “recessive,” it means that one faulty gene copy is not enough to cause disease. Therefore, people can “carry” one faulty SMN1 gene without showing any SMA symptoms; these SMA carriers occur at a rate of about in 1 in 40 people.
When two SMA carriers have a baby, there is a 1-in-4 chance that their child will develop SMA, a 2-in-4 chance that their child will be a carrier, and a 1-in-4 chance that the child will neither have SMA nor be a carrier (Figure 2C).
Can SMA Be Treated?
Like most rare diseases, SMA cannot currently be cured. But three new therapies can improve the symptoms of SMA (Figure 3) [2], and there are several more potential therapies being developed.
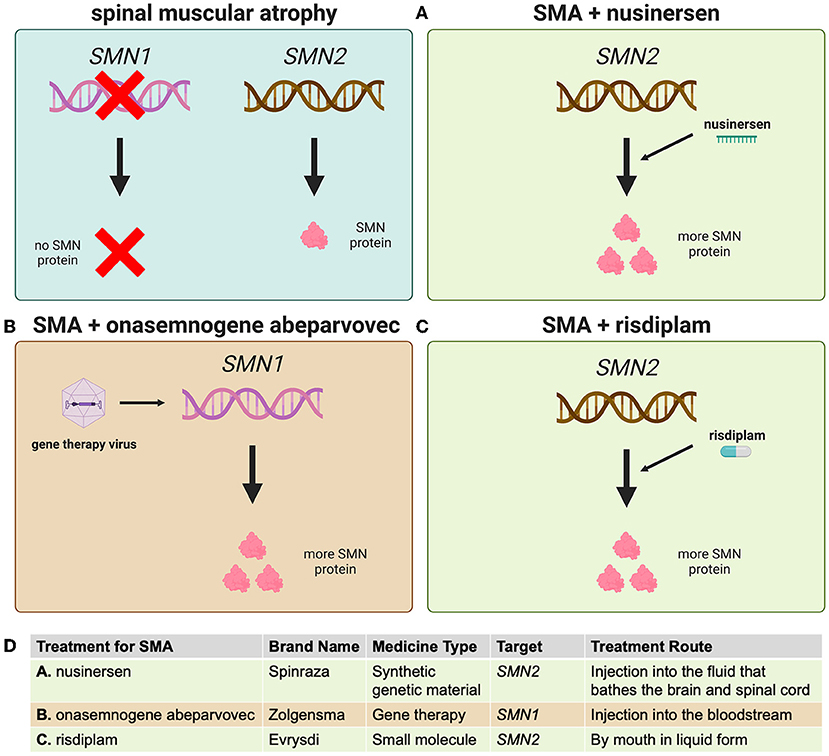
- Figure 3 - Three medicines have been approved to treat the low levels of SMN protein seen in SMA.
- (A) Nusinersen increases the amount of SMN protein made from the SMN2 gene. (B) Onasemnogene abeparvovec is a type of gene therapy that uses a virus to deliver the SMN1 gene to the nervous system. (C) Risdiplam is a small molecule that also increases SMN production from SMN2. (D) A summary of the key features of each approved treatment for SMA.
While we do not know exactly why low SMN levels affect motor neurons, we do know that helping these nerve cells produce more SMN can improve symptoms. This was originally shown in laboratory mice lacking SMN, and it matches what is seen when people with SMA have more copies of the SMN2 gene. The three approved treatments all work by increasing SMN levels in motor neurons (and other cells).
Nusinersen (brand name Spinraza) was the first medicine approved for SMA. This drug specifically recognizes an intermediate molecule made from SMN2 that is used to produce SMN protein, increasing the efficiency with which SMN2 can produce SMN (Figure 3A). Getting medicines into the spinal cord and brain is complicated, as blood vessels and certain cells shield these important organs from potentially harmful substances. So nusinersen must be injected directly into the fluid bathing the spinal cord and brain. Four doses of the medicine are given in the first 2 months of treatment, and then it is administered once every 4 months.
Onasemnogene abeparvovec is a gene therapy known by its brand name Zolgensma. This treatment uses laboratory-modified viruses to carry a copy of SMN1 into cells (Figure 3B). These viruses can enter the spinal cord from the blood and they are also very stable in cells that do not divide, like motor neurons. This means that a single injection of Zolgensma into the blood results in the therapy traveling around the body and entering many kinds of cells, including motor neurons, and then staying active for many years (we do not yet know exactly how long).
The most recently approved SMA treatment, risdiplam (brand name Evrysdi), is a small molecule that works similarly to nusinersen—by increasing SMN production from SMN2 (Figure 3C). Risdiplam is taken daily by mouth in liquid form, and it can also move through the body into many cell types, including motor neurons. This medicine is convenient because it can be taken at home without medical help.
In people with SMA, these three approved treatments all result in more SMN protein in the nervous system, better muscle function, and reduced symptoms. However, none of these therapies can completely cure the disease. Testing indicates that starting therapy even before symptoms begin is the best way to treat SMA. This means babies must be tested for faulty SMN1 genes at birth—something that is being slowly introduced around the world [3]. These three SMA treatments have brought hope to the rare-disease community, showing how improved understanding of a disease can lead to successful therapies that save lives and reduce suffering.
Conclusion: SMA Is a Treatable Disease
A diagnosis of SMA has a profound impact on patients and their families. Fortunately, several treatments have been developed that can reduce the severity of SMA and increase the person’s chance of a healthier life. These treatments have required much time, energy, and money, so we should appreciate the many people involved, including scientific researchers, doctors, patient organizations, drug companies, and, most importantly, SMA patients and their dedicated families. The success of SMA treatments clearly shows the importance of researching rare diseases and demonstrates how a motivated, supportive, and collaborative community is instrumental to developing successful medicines. Hopefully, future work will help us to develop even better treatment strategies for SMA and other rare diseases [4].
Glossary
Atrophy: ↑ The wasting away of a tissue or organ due to lack of use. Muscles atrophy in people with SMA.
Motor Neurons: ↑ Nerve cells that carry electrical signals to muscles to cause them to contract.
Cell Body: ↑ The part of the cell where the nucleus and genetic material are located, considered the command center of the cell.
Axon: ↑ A long, thin, tube-like extension of a nerve cell (neuron) that is vital for quickly sending electrical signals, for example to muscles.
Genetic Condition: ↑ A disease that is inherited through DNA.
Autosomal Recessive: ↑ A way some genetic diseases are passed on to children that requires two faulty gene copies to be inherited (one from each parent) for the disease to occur.
Carrier: ↑ Someone who can pass on a faulty, disease gene to their children, but does not themself have the disease.
Gene Therapy: ↑ A treatment that aims to change a person’s genes or gene activity to treat a disease.
Acknowledgments
The authors would like to thank Kathleen Singleton and Helen A. Sleigh for critical reading of the manuscript. This work was supported by a Medical Research Council Career Development Award (MR/S006990/1; JS) and funding from Spinal Muscular Atrophy UK (SMA Research Consortium; RY-M). All figures were generated using BioRender software (https://Biorender.com/).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
[1] ↑ Mercuri, E., Sumner, C. J., Muntoni, F., Darras, B. T., and Finkel, R. S. 2022. Spinal muscular atrophy. Nat. Rev. Dis. Primers 8:52. doi: 10.1038/s41572-022-00380-8
[2] ↑ Chaytow, H., Faller, K. M. E., Huang, Y. T., and Gillingwater, T. H. 2021. Spinal muscular atrophy: from approved therapies to future therapeutic targets for personalized medicine. Cell Rep. Med. 2:100346. doi: 10.1016/j.xcrm.2021.100346
[3] ↑ Dangouloff, T., Vrščaj, E., Servais, L., Osredkar, D., and SMA NBS World Study Group. 2021. Newborn screening programs for spinal muscular atrophy worldwide: where we stand and where to go. Neuromuscul. Disord. 31:574–82. doi: 10.1016/j.nmd.2021.03.007
[4] ↑ Wirth, B., Karakaya, M., Kye, M. J., and Mendoza-Ferreira, N. 2020. Twenty-five years of spinal muscular atrophy research: from phenotype to genotype to therapy, and what comes next. Annu. Rev. Genomics Hum. Genet. 21:231–61. doi: 10.1146/annurev-genom-102319-103602