
Think about what your brain does when you remember. As an experiment, try to remember what you had for lunch yesterday. Maybe it will come to you immediately, but chances are that you will have to think back about where you were when you were eating lunch, whom you were with, what you were talking about or thinking about, and so on. Eventually, the memory of your lunch will reappear! What you have actually done is recalled a specific event – that is, a time, place, and series of sensory experiences – that together constitute a complex memory. We refer to this type of memory as episodic memory, because it represents a specific episode or event in your life. In general, episodic memory refers to memory for events, and you recall them by performing “mental time travel”; that is, thinking back to where you were and what you were doing to recall elements of the event that allow you to retrieve the entire event. In some cases, this might happen without much effort (say, if you saw a really memorable movie) and in other cases it might require more concentration or effort. But this process is the core of what most people mean when they talk about “memory.” There are other types of memory, though. Semantic memory is memory for facts, like the names of objects or your knowing that water boils at 100°C. Procedural memory refers to memory that you may not be consciously aware of, but which you can demonstrate – riding a bike is one example of procedural memory that reflects motor skills.
Why the fuss about episodic memory? This is the type of memory that is very susceptible to failure in many situations. Memory failure of this sort is usually referred to as amnesia. One of the most common examples of episodic memory failure is experienced by people with Alzheimer’s disease (AD) [1]. AD is actually a type of dementia, which is a much more severe problem than amnesia. Patients with dementia suffer from multiple types of cognitive problems that include amnesia, aphasia (trouble using and understanding language), difficulty with abstract thinking, and trouble performing complex activities. AD usually begins with amnesia that gradually progresses over many years to the point where people who have AD can no longer care for themselves. Although there are some medications that can produce slight improvements in the symptoms of AD, these medications have very small benefits and do not attack the fundamental problems of the disorder.
Within the past 10 years, there have been major advances in understanding the basic biology of AD, and many people are now hopeful that we may develop an effective treatment. There are two major abnormal brain structures that accumulate in the brain in AD. One, called the neurofibrillary tangle (NFT), is made of a protein called tau that accumulates in brain cells (or neurons) (Figure 1A). The other, called the plaque, is made of a protein called β-amyloid (or Aβ) (Figure 1B) that is deposited in the space between neurons. It has long been debated whether one of these proteins or structures might be the initial abnormality, or even the cause of AD. While we do not know the answer, there is considerable evidence that the Aβ plaque is the first event in the development of AD. Much of this evidence comes from genetic causes of AD which, while they are extremely rare, all produce increases in the amounts of Aβ, suggesting that this alone is sufficient to initiate the beginning of AD.
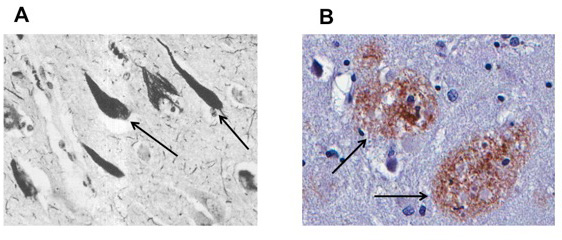
- Figure 1
- A. Arrows point to neurons with tau pathology. These neurons are affected by the paired helical filaments containing pathological accumulations of tau. B. Arrows note β-amyloid plaques, large deposits of the amyloid protein that is characteristic of AD.
One of the early insights into the origins of AD was the observation that the tau NFTs are particularly concentrated in the medial temporal lobe of the brain, in areas that include the entorhinal cortex and hippocampus [2]. These parts of the brain work together as key components of the brain’s episodic memory system. Medial temporal lobe structures seem to be important in binding together different aspects of what we call a memory, so that the memory can later be retrieved. In the example, we started with, when you think about your lunch from yesterday, you may have initially remembered that you were in the cafeteria at school – by the time you recalled the whole episode, you remembered many different aspects of that experience (the lighting, the sounds, the people, and the food). Your hippocampus was involved in binding together all of the different aspects of that experience so that it constituted a single event that you could recall as a memory. If the hippocampus is damaged, memory abilities decline, the damage of the hippocampus on the left side will usually interfere with verbal memory (for things you hear or read) while damage to the hippocampus on the right side will usually interfere with visual memory (things you see). The presence of tau NFTs in the hippocampus and entorhinal cortex likely interferes with the transfer of information into and out of the hippocampus and thus interferes with the ability to encode memories. The hippocampus can be seen on an MRI scan (Figure 2A) and it is atrophic in people with AD and in those who have a progressive but severe amnesia, which we call Mild Cognitive Impairment (MCI). These people are likely in an early stage of AD.
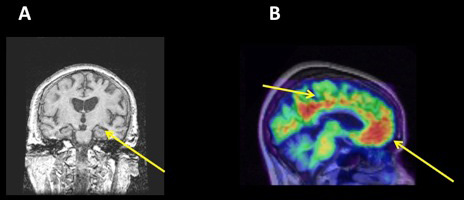
- Figure 2
- A. A coronal view of the brain, taken with magnetic resonance imaging. The arrow points to the medial temporal lobe, which is atrophic in patients with AD. The degree of atrophy is related to accumulation of NFT-tau pathology. B. A PET scan taken using Pittsburgh Compound B, which binds to brain β-amyloid. Areas of hot colors (reds) denoted by the arrows have accumulation of amyloid. These regions are particularly part of the default mode network.
The Aβ plaque affects other parts of the brain than the NFTs do. The plaque is particularly found throughout the cerebral cortex in regions of the frontal, parietal, and temporal lobes that are involved in higher order cognitive function. The Aβ plaque can now be imaged using PET scanning [3]. This technique makes use of radioactivity that is part of a molecule that can be injected into a vein, and which binds to the Aβ plaque in the brain. These scans involve a small injection through a needle, which hurts a little bit, but the scan itself is painless. The molecule provides the binding to the target (Aβ) and the radioactivity allows us to detect this binding using a PET scanner, so that we can make a map of the Aβ in the brain. A particular network of brain structures is affected by the Aβ plaque (Figure 2B). This network is called the “default mode network” (DMN) because in the default state, when you are not focusing on any particular cognitive task but perhaps daydreaming or otherwise engaged in internally driven thinking, this system is active. It “turns off” or becomes less active when you engage in a specific cognitive function like attending an external stimulus of some sort. In particular, in a number of experiments, when people were shown visual stimuli of different types (images, faces, etc.), the DMN became less active, and the more the brain deactivated the better the stimuli were remembered. It is now well recognized that the Aβ is preferentially deposited in the DMN, and that Aβ is associated with less deactivation of the DMN which, in turn, is associated with memory dysfunction.
We now understand two reasons that memory fails in people with AD, NFT deposition in the medial temporal lobe, and Aβ deposition in the DMN. Experiments in older people who are completely healthy (i.e., without AD or amnesia) have shown atrophy in the medial temporal lobe, Aβ in the DMN, and alterations in DMN activity. A number of studies are beginning to see if we can lower Aβ with specific drugs in order to prevent the development of AD in such individuals. These are complicated and expensive studies, but for the first time, they offer hope of prevention of this disease. It will be important to study these older people who have signs of NFT and Aβ in the brain but who do not have symptoms, so that we can treat them before they get sick. At this point, we do not know how to repair brains that have been damaged, so treating before the damage occurs offers the best hope of treating AD.
References
[1] ↑ Ballard, C., Gauthier, S., Corbett, A., Brayne, C., Aarsland, D., and Jones, E. 2011. Alzheimer’s disease. Lancet 377:1019–31. doi: 10.1016/S0140-6736(10)61349-9
[2] ↑ Braak, H., and Braak, E. 1997. Frequency of stages of Alzheimer-related lesions in different age categories. Neurobiol. Aging 18:351–7. doi: 10.1016/S0197-4580(97)00056-0
[3] ↑ Rabinovici, G. D., and Jagust, W. J. 2009. Amyloid imaging in aging and dementia: testing the amyloid hypothesis in vivo. Behav. Neurol. 21:117–28. doi: 10.3233/BEN-2009-0232