
Abstract
When I was young, I wanted to be an astronaut and I was a hard-core science fiction fan. I was always drawn to heroic characters that invented something new that resulted in an amazing breakthrough. When I started my scientific career, I did not want to do something small—I wanted to do something that would be really different and impactful. This is why I chose to work on one of the most difficult problems in the field of optical (light-using) microscopy—how to see objects that are smaller than the wavelength of visible light. This meant challenging a limit that was long believed to be unbreakable. In my research, I developed a method called photoactivated localization microscopy (PALM), which enabled us to break that limit with the help of glowing (fluorescent) molecules. Using the PALM method, and other methods based on glowing molecules, scientists can learn new things about living cells and single molecules and significantly advance our understanding of life.
Professor Eric Betzig won the Nobel Prize in Chemistry in 2014, jointly with Prof. Stefan Hell and Prof. William Moerner, for the development of super-resolved fluorescence microscopy.
How Can We See Small Things?
When we look at any object, we are actually detecting the light that bounces back from the object and reaches our eyes. Think, for example, about a submarine using sonar. When a submarine is navigating in the sea, it sends out sound waves and detects the waves that bounce back to it from underwater objects, such as rocks and sea animals (Figure 1A). In this way, the crew of the submarine knows how to navigate underwater. The same principle applies when we want to see things in the laboratory, such as cells or tiny organisms. We shine light (or some other form of radiation, such as electrons) on the tiny object, and we look at what bounces back from the object. The type of radiation is determined by a measure called the wavelength. As you might know, waves have a repeating pattern of high peaks and low troughs. The wavelength is the distance between two peaks (Figure 1B).
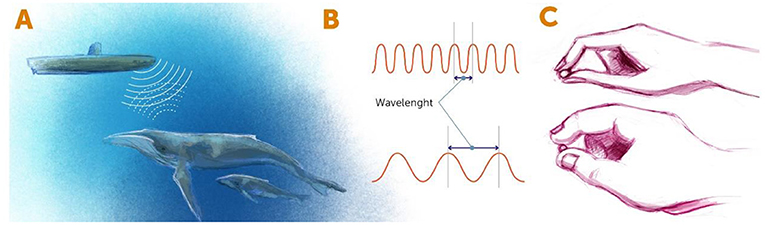
- Figure 1 - Detecting objects using radiation.
- (A) In the sonar systems of submarines, radiation of sound waves is used to detect nearby objects. The sound waves are emitted from the submarine, bounce off nearby objects, and return to the detector in the sonar system. Similarly, when we see something with our eyes, light waves hit the object and bounce back to our detectors (our eyes). (B) Forms of radiation can be described by their wavelengths. Wavelength is defined as the distance between two adjacent peaks of the radiation wave. (C) A short wavelength (top) is like small fingers that help us “see” the small details of an object, whereas a long wavelength (bottom) is like chubby fingers, with which we can “see” only obvious details.
In terms of looking at tiny objects, the wavelength of energy bouncing off that object determines the resolution with which we can see that object. The shorter the wavelength is, the smaller the objects we can see. You can think about it like fingers that are trying to “feel” an object (Figure 1C). If the fingers are thick (meaning long wavelengths), we cannot sense the fine details of the object—it is like trying to feel millimeter-wide features with chubby fingers. So, if we want to see the tiny details of an object, we have two choices. First, we could use a form of radiation with a very small wavelength, such as x-rays. The problem is that prolonged exposure to the high energy of short wavelengths can kill living things, so we cannot study living cells or organisms using such short wavelengths. The other choice is to find some trick that will enable us to use longer wavelengths, which are less energetic, while somehow managing to see beyond the limits of those wavelengths. This is the idea behind super-resolution microscopy, which is the term for any method of microscopy that allow us to see beyond the limits of the wavelengths that it uses.
Before the development of super-resolution microscopy, we could see living things as small as about 200 nanometers (or 0.0002 millimeters)—such as the larger compartments within the cells of animals, and even one-celled organisms like bacteria. We could not see smaller organisms such as viruses, or smaller parts of cells like individual proteins or other small molecules (Figures 2A, B). The ability to see living things at such high resolution was a huge leap forward! It opened up a whole new area of research and gave researchers the potential to better understand the most fundamental processes of life. Details that were previously undetectable using conventional microscopy techniques were exposed right before our eyes, and generated great excitement for studying the mysteries of life (Figures 3A, B).
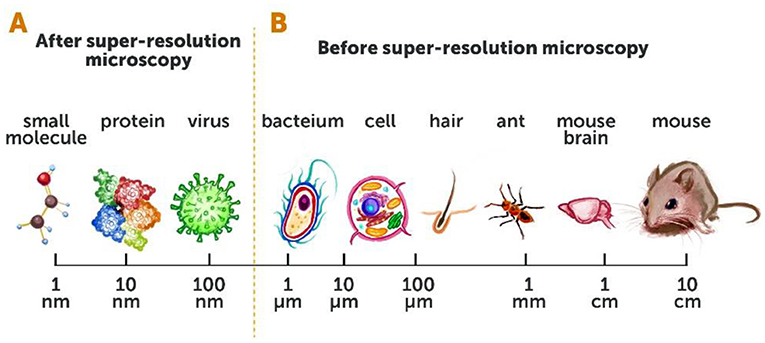
- Figure 2 - Conventional microscopy vs. super-resolution microscopy.
- (A) Super-resolution microscopy has opened up our ability to see a whole range of objects, down to ~10 nanometers [1 nanometer (nm) = one billionth (0.000000001) of a meter]. (B) Before super-resolution microscopy, we could only view objects that were 200 nm in size (about the size of a bacterium) or larger.
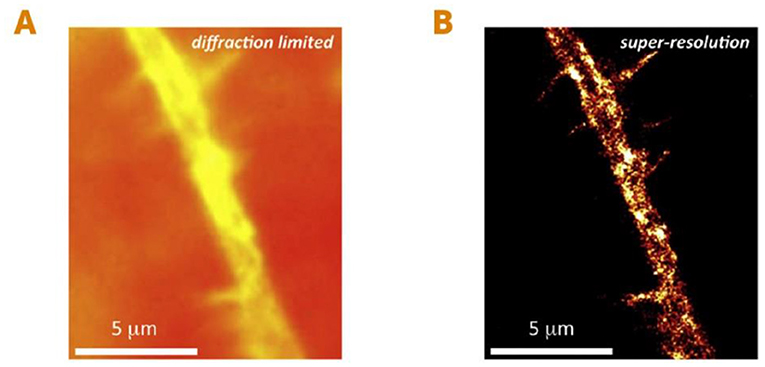
- Figure 3 - Seeing individual proteins in living cells using super-resolution microscopy.
- (A) An image of a small branch of a living nerve cell, taken using conventional microscopy. (B) The same nerve cell branch using super-resolution microscopy. This method allows us to see small details that were previously unobservable. In this case, we can see small ion channels, which are proteins in the nerve cell’s membrane (bright yellow spots) responsible for conducting electricity in nerve cells [figure adapted from [1]]. Scale: one micrometer (μm) is 1,000 nanometers.
The Beginning: Near-Field Microscopy
When I started my advanced studies at university in 1983, two of my professors—Mike Isaacson and Aaron Lewis—had the crazy idea of trying to break what was called the Abbe’s diffraction limit. This was the concept that the smallest thing we can see using light waves must be at least half the wavelength of that light. For example, if the wavelength is 1 mm, we could see objects that are at least 0.5 mm long. My professors thought they could break this limit by manipulating light in a specific way. Their idea was based on the first demonstration of breaking the Abbe’s diffraction limit, which was performed back in 1972 [2]. The basic idea was to drill a tiny hole, much smaller than the wavelength of light, in a small black plate. When the plate is placed very close to the object to be examined, and a light is shone through the hole, it illuminates a very tiny spot of the object—much smaller than the wavelength of light. The object can then be “scanned” by moving the illuminated plate around, point by point, across the object. Using this trick, we can see the object with resolution higher than the “natural” resolution of the incoming light. Today, this method is called near-field scanning optical microscopy [3, 4].
This was the first super-resolution microscopy method that I worked on. The main problem with this method is that the light that passes through the small hole spreads out very quickly on the other side. To get high resolution, we must work extremely close to the object we are imaging. In the case of cells, for example, this is challenging because cells are not flat, so it is difficult to control the plate on top of them. After working on this method for several years and taking it as far as I thought possible, I decided to quit this work, and science altogether. Little did I know that, a few years later, a big breakthrough in the field of biochemistry would bring me rushing back to science and microscopy.
Super-Resolution Fluorescence Microscopy
In 1994, a pioneering study was published [5] showing that, using genetic engineering, we can attach a glowing handle, or “marker,” called a fluorescent protein, to any protein in living cells. This is a special protein that glows when one specific wavelength of light is shone on it. I immediately realized that this work was a complete game changer in the field of microscopy, because it could help us to see the tiny structures of inside of cells. A year later, in 1995, I published a paper that laid out the foundations for a new method of microscopy [6]. But it was only in the early 2000s that additional advancements in the field of fluorescent molecules allowed me to follow through on my idea. The advancement created molecules that were not always fluorescent, but could be “activated” to glow when a certain wavelength of light was shone on them [7]. This meant that we could attach glowing markers to certain proteins inside living cells and intentionally activate them, to study cellular structures and processes. That was the start of the method of super-resolution fluorescence microscopy that I helped to develop, which was originally called photoactivated localization microscopy (PALM) [8, 9]. In 2014, I received the Nobel Prize in Chemistry for this method.
The idea behind PALM is as follows: each cell contains about 20,000 different kinds of proteins, and often many thousands of each kind. We want to understand how they work together. Using a conventional microscope like you have in biology class at school, all you can see when you look at these proteins in a cell is a big glowing blob. The proteins are so close together that you cannot tell them apart. In PALM, we attach special fluorescent markers to proteins—markers that can be turned on by a low-power (violet light) laser (Figure 4, step 1) and then glow and become detectable when a separate, higher-power (blue light) laser is shone on them (Figure 4, step 2).
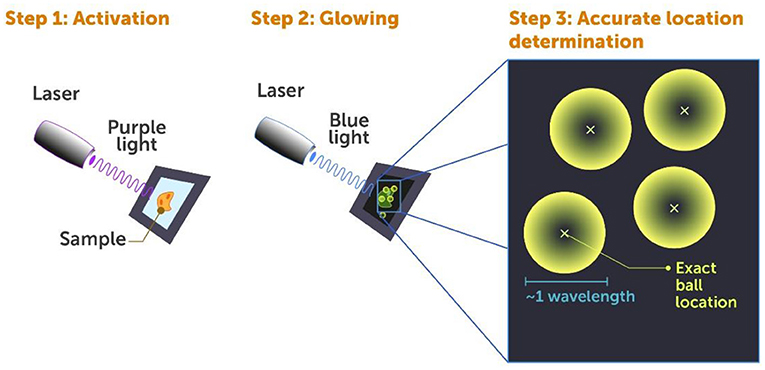
- Figure 4 - Photoactivated localization microscopy (PALM).
- Super-resolution microscopy of fluorescently marked cells using the PALM method includes three steps: Step 1: A laser beam of weak violet light is shone on the cell in brief pulses, to activate the fluorescent markers on only some of the proteins, making them ready to glow. Step 2: A blue laser beam is shone on the cell, which makes the activated proteins glow so they can be detected. Step 3: The location of individual proteins is determined by a computer, by finding the center (small x) of each “ball of light” generated by each fluorescent protein.
If we were to activate all the markers at once, they would all glow at the same time and this would create a big mess—that is where the big glowing blob comes from. This is why we use pulses of very low energy violet light to activate the markers—only a few at a time with each pulse. These few are randomly activated and are most likely well-separated from one another within the cell. When we then use blue light to detect these activated markers, they look like small glowing balls (Figure 4, step 2) ~1 wavelength of light in size, since that is the smallest anything can appear in the conventional diffraction-limited microscope we use to look at them (Figure 4, step 3).
Now here is the trick—we can use a computer to process the image we get from the microscope and accurately find the center of each of these “balls”. You can think of it like a basketball which had a spherical shape with a certain diameter. You are able to point to the very center of the basketball with much better precision than estimating its diameter, even if you do not directly see the center. The same is true for these molecular balls—we can find their centers with very high precision, much closer to their true size than the size of the glowing balls. This means that, each time we pulse the cells with light, we can find the positions of a small set of proteins within the cell (Figure 4, step 3). The fluorescence of these proteins turns off naturally; then we can illuminate another set of proteins and find their location. Typically, it takes tens of thousands of rounds of activation to map a whole cell. But the effort is worth it, as we get a very high-resolution image of the cell or any other object we are studying (see Figure 3B and images in Betzig et al. [9]).
Challenges and Potentials of Super-Resolution Microscopy
As you have seen, PALM is quite simple—all you need is a laser to shine a beam of light on the object, a camera, and a relatively simple software program to find the centers of the glowing proteins. This equipment is cheap and simple. In fact, my friend Prof. Harald Hess and I built the first model of PALM in his living room, with equipment that we bought with our own money while we were both unemployed! The difficult part involves working with the biological sample. There are many challenges including difficulties preparing living cells for our experiments, causing damage to the cells in response to the light, and figuring out the best way to detect and analyze the light emitted by the molecules we are interested in.
In terms preparing the cells, it turns out that many of the markers we can activate using light do not actually attach to the proteins that we want to see, but rather to other objects that happen to be nearby. This means that often the markers we use do not actually point us toward the locations of the proteins we are interested in. Additionally, even if we manage to label the correct proteins, we label only a small percentage of them—which is often not enough to give us a full image of the cell at the highest possible resolution. Even if we manage to label enough of the correct proteins, cells do not like to have intense light shone on them. Yet, the more intense the light we shine on the cells, the more information we can get. Therefore, we are always trying to find the balance between extracting as much information as possible information while avoiding damage to the cells.
The last challenge that I will mention here is a phenomenon called photobleaching. Photobleaching describes the fact that a marker can only glow a certain number of times. In other words, only a limited amount of light can radiate from a specific marker before the marker is destroyed or becomes permanently dark. Sometimes, this amount of light is not enough for us to extract the information we need to find the marker’s exact location.
As I mentioned previously, super-resolution fluorescence microscopy is unique because it enables us to image live cells and organisms. Using this technique, we do more than simply determine the structure of living things—we can also track processes happening within a cell, such as the movement of proteins, over time (see this video) [10, 11]. Using what is called single-molecule tracking, we can see into the deepest mysteries of living cells and have a peek into the most fundamental processes of life. For example, single-molecule tracking has helped us to understand how RNA copies are made from the DNA inside a cell’s nucleus—a process called transcription).
Tracking single molecules and figuring out how they move around within cells might be very important for the development of new drugs. In my view, the information we can learn about cellular mechanisms that were previously invisible to us might lead to a whole new paradigm of drug discovery and to important new treatments for various diseases, such as Alzheimer’s and Parkinson’s diseases. I think this might be the greatest thing that will come out of super-resolution microscopy, and it is why my colleagues and I started a drug-discovery company called Eikon Therapeutics©.
Recommendations for Young Minds
As I mentioned earlier, when I was a kid, heroic fantasy characters and astronauts inspired me. They represented the chance of having an impact on the world, by significantly improving people’s lives. For me, that is the highest goal a person can choose in life. Therefore, I recommend that, whatever you do in your career, do something impactful that provides a meaningful contribution. It does not have to be big thing—raising children is impactful, and packing groceries is also impactful. Try to find something that is a good mixture of your own interests and that also has the potential to positively impact the people around you or society as a whole (Figure 5). If you do choose to become a scientist, do not get stuck on the idea of becoming a professor. This should not be a goal in itself, since there are many other ways to make meaningful contributions and great discoveries outside of working at a university.

- Figure 5 - Recommendations for Young Minds.
- As you build your future, try to do something that you like and that also provides a meaningful contribution to society.
Personally, I find many advantages to doing the kind of research that I do. First, I am my own boss. I enjoy that because I like to make decisions myself rather than being told what to do. Second, in the type of science that I do, I try to invent new tools for people who are trying to answer scientific questions that are outside my expertise. This means I must learn lots of new things and become a “jack of all trades”. I know a little bit about many things in many areas—from which materials work best in different machines, to biology and physics, to designing new research tools. This broad knowledge spills over into my day-to-day life, and I now understand things I see around me and enjoy the beauty and complexity in my everyday world.
The last thing I would like to discuss is the attitude you choose in whatever you do. First, you should never forget to think critically about any question that you are faced with. Do not be satisfied with superficial, automatic thinking—really try to see into the depths of the things you encounter. Second, do not be afraid to take risks. In my opinion, society has become too risk-adverse, which limits our ability to innovate and advance—both as individuals and as a society. Last, hard work is important! No matter what you do, at any age, try to push yourself while also having fun. Do not beat yourself up if things are difficult, but make serious efforts to get what you want. If you do not excel that is OK, because everybody is good at different things—nobody is good at everything. But push hard in everything that you do—hard work will get you anywhere. Find the thing that you love to do, work hard and become good at it, and then run with that ball, make good use of it, and enjoy the process, too.
Glossary
Radiation: ↑ Energy, in the form of waves or particles, that is emitted from a source.
Wavelength: ↑ A measure of the distance between two adjacent peaks of a wave.
Super-resolution Microscopy: ↑ Any method of microscopy which enables us to overcome the limits of the wavelengths it uses and therefore see objects with greater resolution.
Abbe’s Diffraction Limit: ↑ The physical limit of light microscopes whereby we can distinguish between two points in an object only if the distance between them is not smaller than half the wavelength of the imaging light.
Near-field Scanning Optical Microscopy: ↑ The first super-resolution microscopy method that was developed during the 1980s.
Photoactivated Localization Microscopy (PALM): ↑ A super-resolution microscopy method that I developed, that uses fluorescent molecules to break Abbe’s diffraction limit.
Photobleaching: ↑ A phenomenon in fluorescent materials, in which they become permanently unable to shine light after they have done so a certain number of times.
Conflict of Interest
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
I wish to thank Noa Segev for conducting the interview which served as the basis for this paper, and for co-authoring the paper, and Alex Bernstein for providing the figures.
Additional Materials
- Eric Betzig and Harald Hess (Janelia Farm/HHMI): Developing PALM Microscopy.
- Prof. Betzig Nobel Lecture.
References
[1] ↑ Ondrus, A. E., Hsiao-lu, D. L., Iwanaga, S., Parsons, W. H., Andresen, B. M., Moerner, W. E., et al. 2012. Fluorescent saxitoxins for live cell imaging of single voltage-gated sodium ion channels beyond the optical diffraction limit. Chem. Biol. 19:902–12. doi: 10.1016/j.chembiol.2012.05.021
[2] ↑ Ash, E. A., and Nicholls, G. 1972. Super-resolution aperture scanning microscope. Nature 237:510–2. doi: 10.1038/237510a0
[3] ↑ Betzig, E., Harootunian, A., Lewis, A., and Isaacson, M. 1986. Near-field diffraction by a slit: implications for superresolution microscopy. Appl. Opt. 25:1890–900. doi: 10.1364/AO.25.001890
[4] ↑ Betzig, E., and Chichester, R. J. 1993. Single molecules observed by near-field scanning optical microscopy. Science 262:1422–5. doi: 10.1126/science.262.5138.1422
[5] ↑ Chalfie, M., Tu, Y., Euskirchen, G., Ward, W. W., and Prasher, D. C. 1994. Green fluorescent protein as a marker for gene expression. Science 263:802–5. doi: 10.1126/science.8303295
[6] ↑ Betzig, E. 1995. Proposed method for molecular optical imaging. Opt. Lett. 20:237–9. doi: 10.1364/OL.20.000237
[7] ↑ Patterson, G., and Lippincott-Schwartz, J. 2002. A photoactivatable GFP for selective photolabeling of proteins and cells. Science 297:1873–1877. doi: 10.1126/science.1074952
[8] ↑ Shroff, H., White, H., and Betzig, E. 2013. Photoactivated localization microscopy (PALM) of adhesion complexes. Curr. Protocol. Cell Biol. 58:4–21. doi: 10.1002/0471143030.cb0421s58
[9] ↑ Betzig, E., Patterson, G. H., Sougrat, R., Lindwasser, O. W., Olenych, S., Bonifacino, J. S., et al. 2006. Imaging intracellular fluorescent proteins at nanometer resolution. Science 313:1642–5. doi: 10.1126/science.1127344
[10] ↑ Liu, Z., Lavis, L. D., and Betzig, E. 2015. Imaging live-cell dynamics and structure at the single-molecule level. Mol. Cell. 58:644–59. doi: 10.1016/j.molcel.2015.02.033
[11] ↑ Li, D., Shao, L., Chen, B. C., Zhang, X., Zhang, M., Moses, B., et al. 2015. Extended-resolution structured illumination imaging of endocytic and cytoskeletal dynamics. Science 349:aab3500. doi: 10.1126/science.aab3500